青光眼是什么?
大卫Križaj
摘要
青光眼是世界上导致失明的主要原因之一,由于症状通常出现在疾病的晚期,所以诊断起来很困难,而且由于视网膜神经元的不可逆丧失,治疗起来也很困难。该术语包含了以前后眼生物力学改变为特征的一组异质性疾病。这些疾病往往表现为小梁网硬化,房水分泌增加/引流减少,视网膜炎症与激活的小胶质细胞、Müller细胞和星形胶质细胞有关,视网膜神经节细胞变性。一般来说,表型是由诸如年龄、家族史、民族出身、高度近视、血管疾病和眼压(IOP)等危险因素共同引起的。目前的治疗仅限于降低和稳定眼压,这表明青光眼主要是一种眼机械转导疾病。虽然导致眼压升高到神经节细胞损伤的事件还没有很好地定义,但最近的研究表明,机械转导TRPV4、piezo和TREK-1通道和免疫机制是小梁网、睫状体、视网膜神经节、内皮细胞和胶质细胞的关键压力靶点。资料显示,神经节细胞胞体-树突、Müller细胞和小胶质细胞内的机械转导机制功能障碍可能发生在轴突变性、视神经头拔火罐和视野损伤之前,多年来这些被认为是青光眼诊断的金标准。总的来说,这些最近的研究预测,新疗法和诊断策略的发展将取决于对感知和转导眼睛压力的分子机制的系统描述。因此,神经保护这一目前难以实现的目标可能通过同时抑制前眼、视网膜神经元、血管和胶质细胞的压力感知来实现,以实现神经元-胶质细胞-血管相互作用的正常化,并由此降低免疫激活。
简介
青光眼是一组视神经疾病,其特征是视网膜神经节细胞(RGCs)的进行性退变,是发达国家不可逆失明的主要原因。典型症状包括逐渐丧失周围(侧)视力,随后是逐渐丧失中心视力。如果不及时治疗,青光眼可能发展成完全失明。由于对眼睛的损害是缓慢和无痛的,只有一半的携带者意识到这种疾病,而且往往在诊断之前很久就发生了不可挽回的伤害。青光眼通常通过眼压(IOP)调节异常和/或眼部细胞的病理机械敏感性来诊断,但机械力与疾病之间的关系仍然是一个谜,是一个相当大的学术、临床和经济兴趣的问题。青光眼致盲的比例差异很大,从南亚的最低值到撒哈拉以南非洲的高流行率(1)。到2020年,该疾病预计将影响7600万人,估计仅在美国就有300万人患病。目前还没有治愈的方法,因为死亡的视网膜神经元不能再生,然而,通过降低眼压(IOP)的药物可以减缓疾病的进展。考虑到与生产力损失相关的间接成本、身体后果(髋部骨折增加;家庭护理的增加)和生活质量的下降。鉴于青光眼的早期发现、开始治疗和坚持治疗与社会经济地位密切相关(2),在提供护理方面还有很大的改进空间。
最早对类似青光眼的眼疾的描述来自古希腊。亚里士多德和希波克拉底(约公元前500年)使用术语青光眼(γλαυκóς)来描述一种晚期疾病,其特征是瞳孔呈灰绿色-蓝色色调(3)。盖伦(约公元126 -199年)与近角型青光眼的一个显著近似,将这种青光眼色调与房水不足、浅前房和/或前突出晶状体联系起来。10世纪阿拉伯医生阿尔-塔巴里的《希波克拉底治疗之书》青光眼可能提供了一些关于眼压的最早参考,但中世纪的欧洲继续将青光眼归类为一种无法治愈的绿色(绿色)白内障(“瀑布”)和晶状体“硬度过高”(4)。当不透明晶状体被证实可以改善视力时,人们产生了困惑,但与眼压的联系在19世纪中期被Franciscus Cornelius Donders(荷兰)复兴,他开发了第一个眼压计。Albrecht von Graefe(德国)和Antoine-Pierre Demours(法国)将青光眼与虹膜角膜功能障碍联系起来,但是William Mackenzie(苏格兰)在他的《眼科疾病实用论著》(1853)(5)中对青光眼进行了定义和命名。青光眼在中世纪被称为“绿色白内障”,在一些语言(如斯洛文尼亚语)中仍然存在。
青光眼的一个显著特征是发生在眼球内两个不同位置的生物过程之间的必然联系(图1)前的眼睛通过尚不清楚的机制,促成了视网膜视神经病变后眼(视网膜)。为了适应增加的机械力,前眼的小梁网细胞、视网膜神经节细胞和胶质细胞发生结构和功能上的变化,称为青光眼重塑。这一过程包括了纳米尺度(基因表达)、中尺度(酶、离子通道、细胞骨架蛋白、受体和分泌分子的变化)到宏观尺度(神经病变)的许多机制,最终的共同后果是失去对大脑的视网膜输出(图1)。
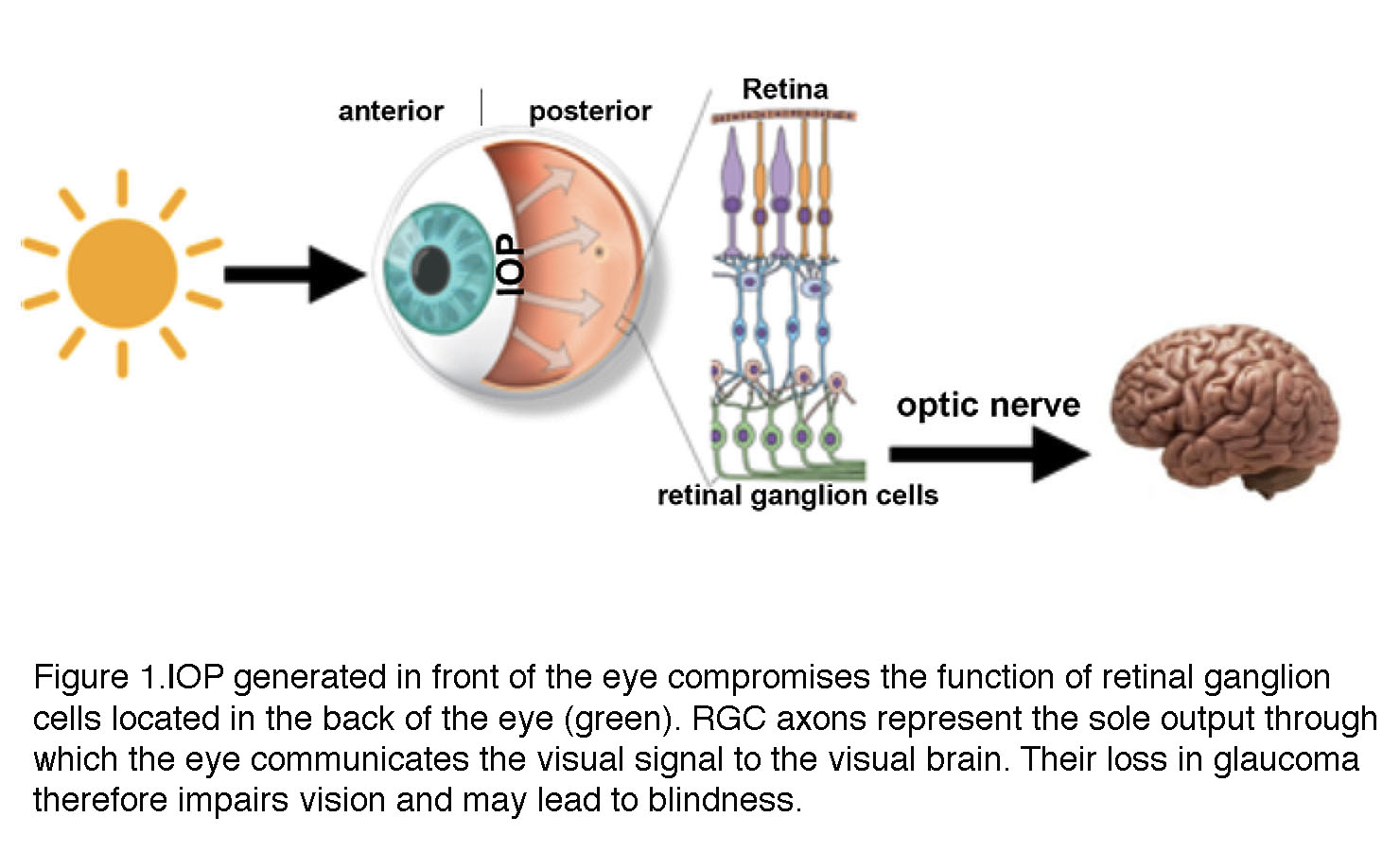
图1所示。眼前(前)产生的眼压损害了位于眼后(后)的视网膜神经节细胞(绿色,RGC)的功能。RGC轴突通过视神经代表唯一的输出,通过眼睛将视觉信号传递到大脑的视觉区域。因此,青光眼的轴突丢失会损害视力,并可能导致失明。
在影响青光眼发生可能性的诸多危险因素中,有年龄、种族、家族史、角膜厚度、低血压、脑脊液压、眼压和血管失调。这些压力源的多样性,其中一些是独立于眼压的,是青光眼进展率和对治疗反应在个体患者之间的差异的基础。
值得注意的是,青光眼的诊断标准还不能确定青光眼的分子和生理标记在青光眼的诊断和治疗中,缺乏一个将青光眼危险因素和表型相结合的结构-功能框架是一个重大的未满足的需求.教科书上广泛使用的青光眼定义是视神经病变,定义为视神经头的“拔火罐”是一种过时的对一种晚期疾病的描述,在这种情况下,IOP诱导的视网膜神经元兴奋性、突触重构、神经炎症和光反应性改变的早期预警阶段已经发生。它也未能洞察正在进行的争论,青光眼是一种表型连续的疾病,还是一组具有不同遗传起源和信号传导机制的疾病。这篇综述提供了当前一些有争议的话题和/或目前缺乏知识的领域的鸟瞰概述。这反映了作者的个人观点,也遗憾的是,绝大多数相关研究不能直接引用(虽然有很多在二手文献中引用)。
青光眼的诊断。
目前青光眼的临床诊断是基于IOP、视觉功能和视网膜结构的测量。立体“裂隙灯”视镜而且光学相干断层扫描(OCT)检查视神经头“杯状”(凹陷)及视网膜神经纤维层(由RGC轴突组成)退变;视野测量通过弧形盲点(周边视力的局部丧失)的检测记录视力的功能性损伤张力测定法用于测量眼压(6,7)。有原发性闭角型疾病风险的患者(见下文)还需要用前房角镜检查它通过一种特殊的晶状体将光线反射进眼睛。眼内表面(“眼底”)的眼镜检查示例如图2和图3所示。“挖出”或“杯状”的视神经头图2与中心凹区血管化减少有关,而图3显示了一个典型的晚期例子,其中有一个正常患者视网膜视神经头的放大视图(左)和一个严重青光眼患者视网膜的相同视图(右)。
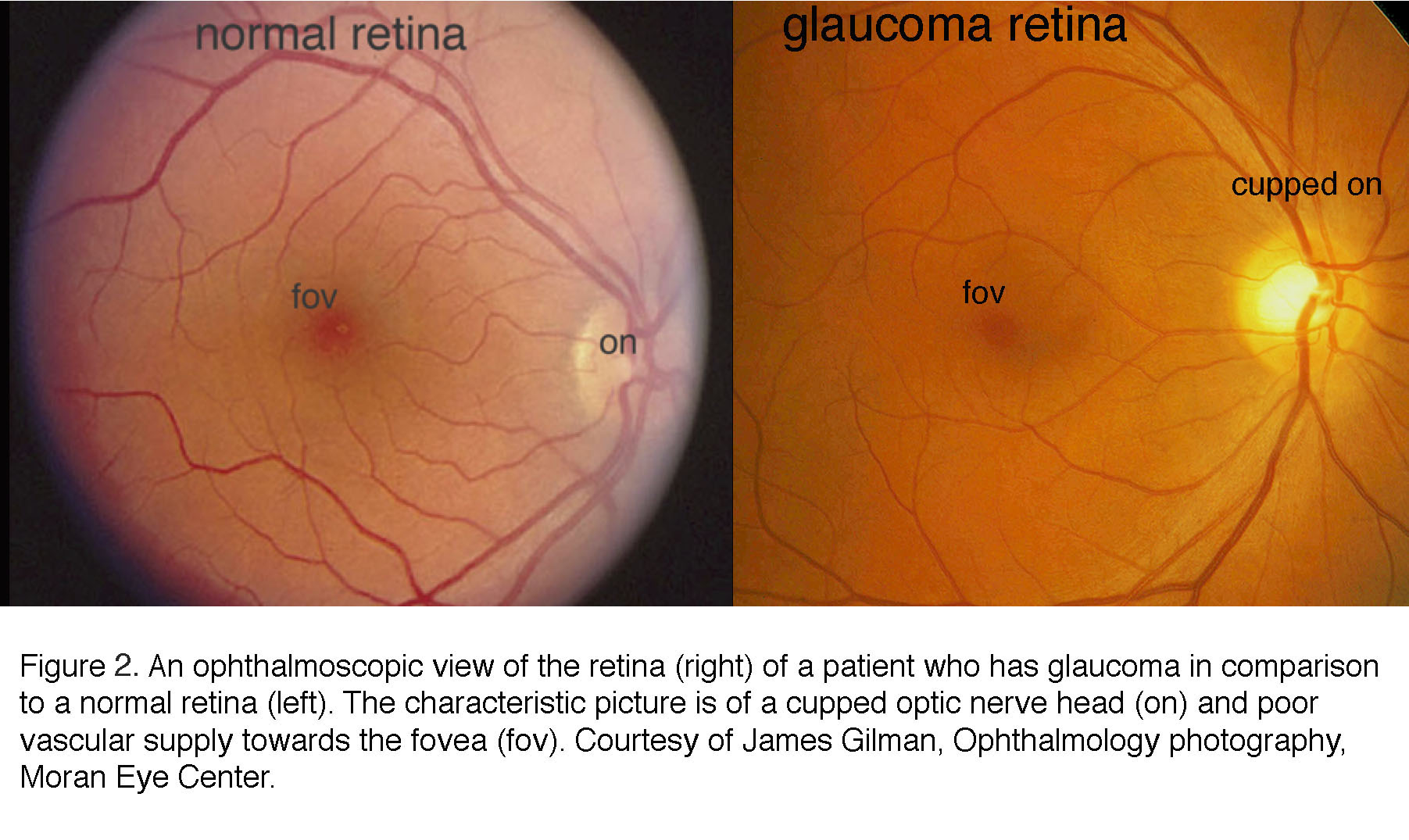 图2。青光眼患者的视网膜(右)与正常视网膜(左)的对比。典型的图像是杯状视神经头(上)和血管向中央凹(fov)的供应不足。由詹姆斯·吉尔曼提供,眼科摄影,莫兰眼科中心。
图2。青光眼患者的视网膜(右)与正常视网膜(左)的对比。典型的图像是杯状视神经头(上)和血管向中央凹(fov)的供应不足。由詹姆斯·吉尔曼提供,眼科摄影,莫兰眼科中心。
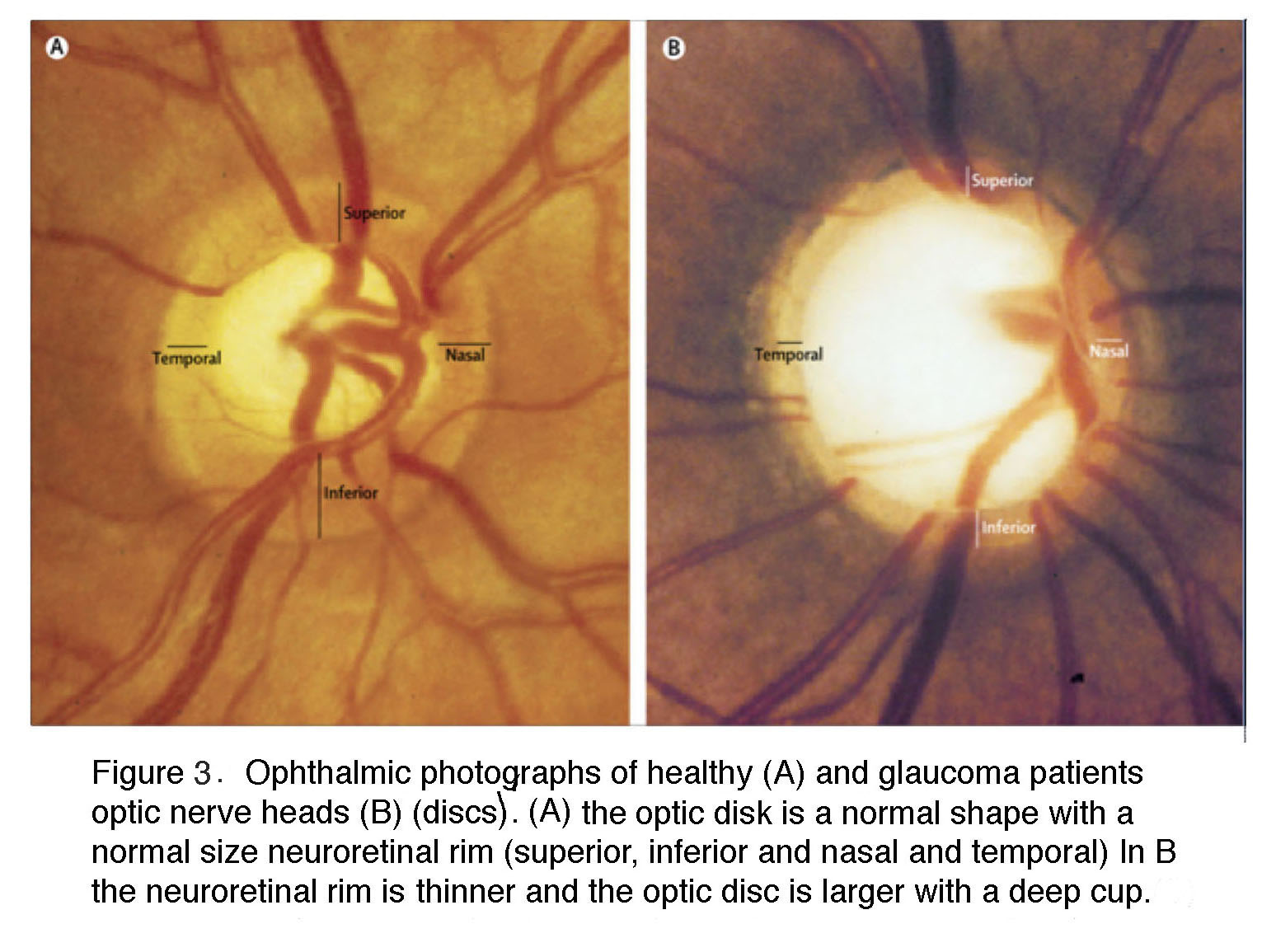
图3。健康人(A)和青光眼患者视盘(B)的眼科照片。视盘是视神经头。(A)视盘形状正常,视网膜神经边缘大小正常(上、下、鼻和颞)。(B)视网膜神经缘较薄,视盘较大,有深杯。
在青光眼中,视神经头显得较大,因为神经视网膜边缘较窄,中心呈杯状。在晚期青光眼的眼底照片中,神经节细胞轴突退行性变可以通过剩余轴突的“楔形”外观看到,这与健康的神经节细胞轴突向视神经头的上、下弓辐射不同(图4)。
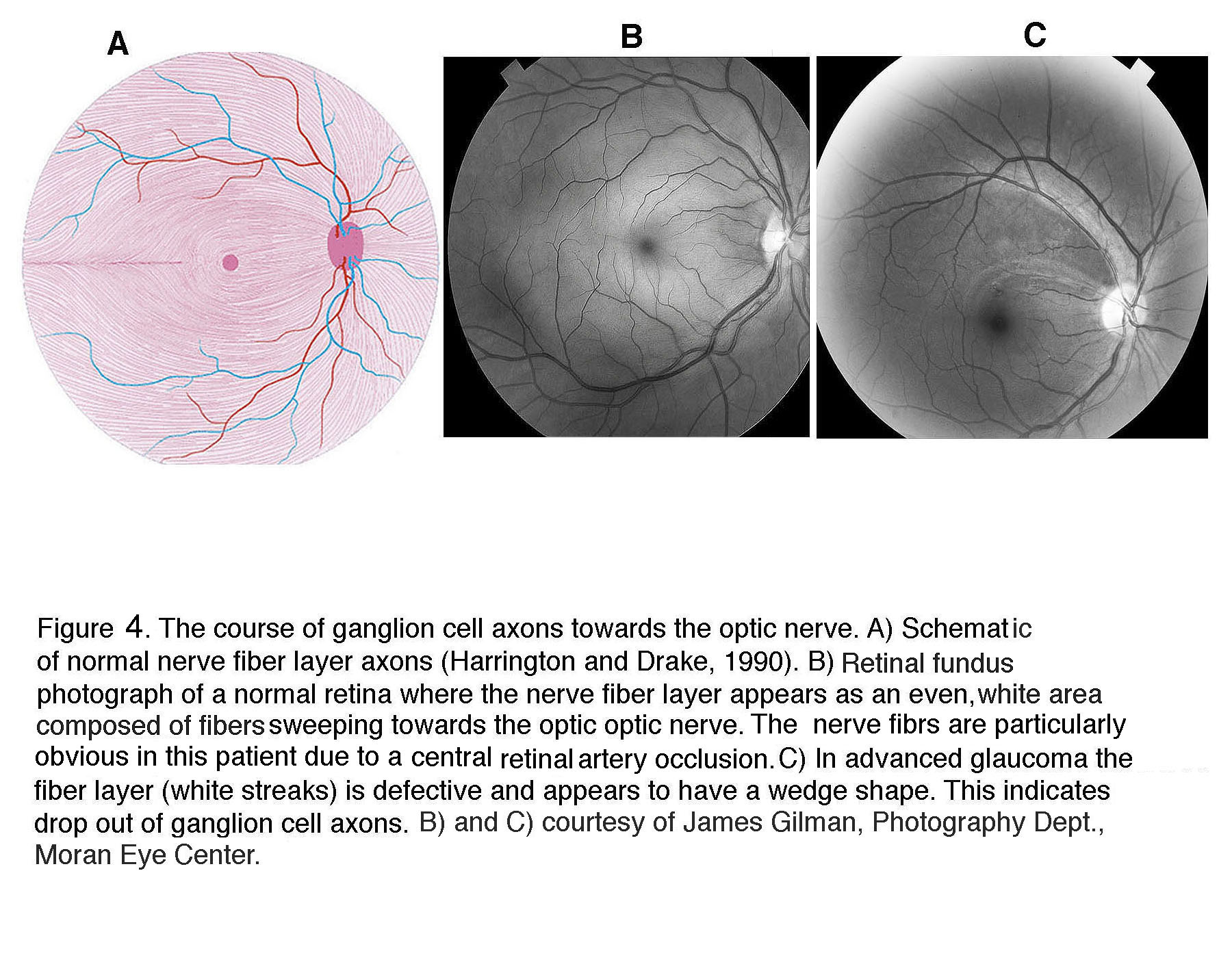 图4。神经节细胞轴突走向视神经头的过程。(A)正常神经纤维层轴突示意图(Harrington and Drake, 1990)。(B)正常视网膜眼底照片,神经纤维层呈均匀的白色区域,由向视神经头延伸的纤维组成。由于视网膜中央动脉阻塞,该患者的神经纤维特别明显。(C)在晚期青光眼中,纤维层(白色条纹)有缺陷,呈楔形。这表明神经节细胞轴突脱落。(B)和(C)由詹姆斯·吉尔曼提供,摄影系,莫兰眼科中心。
图4。神经节细胞轴突走向视神经头的过程。(A)正常神经纤维层轴突示意图(Harrington and Drake, 1990)。(B)正常视网膜眼底照片,神经纤维层呈均匀的白色区域,由向视神经头延伸的纤维组成。由于视网膜中央动脉阻塞,该患者的神经纤维特别明显。(C)在晚期青光眼中,纤维层(白色条纹)有缺陷,呈楔形。这表明神经节细胞轴突脱落。(B)和(C)由詹姆斯·吉尔曼提供,摄影系,莫兰眼科中心。
一般来说,一旦30%的rgc死亡,就可以检测到功能丧失,尽管患者和动物可能直到很久以后才会注意到他们的视力受损。图5显示RGCs是诱发青光眼的非人灵长类动物视网膜中丢失的主要神经元细胞类型。患有诱导性青光眼的猕猴视网膜(8)在慢性眼压升高后显示RGCs大量丢失(图5A),但只有在猕猴失去约50%的RGCs后才显示视觉敏感性降低(图5B)。
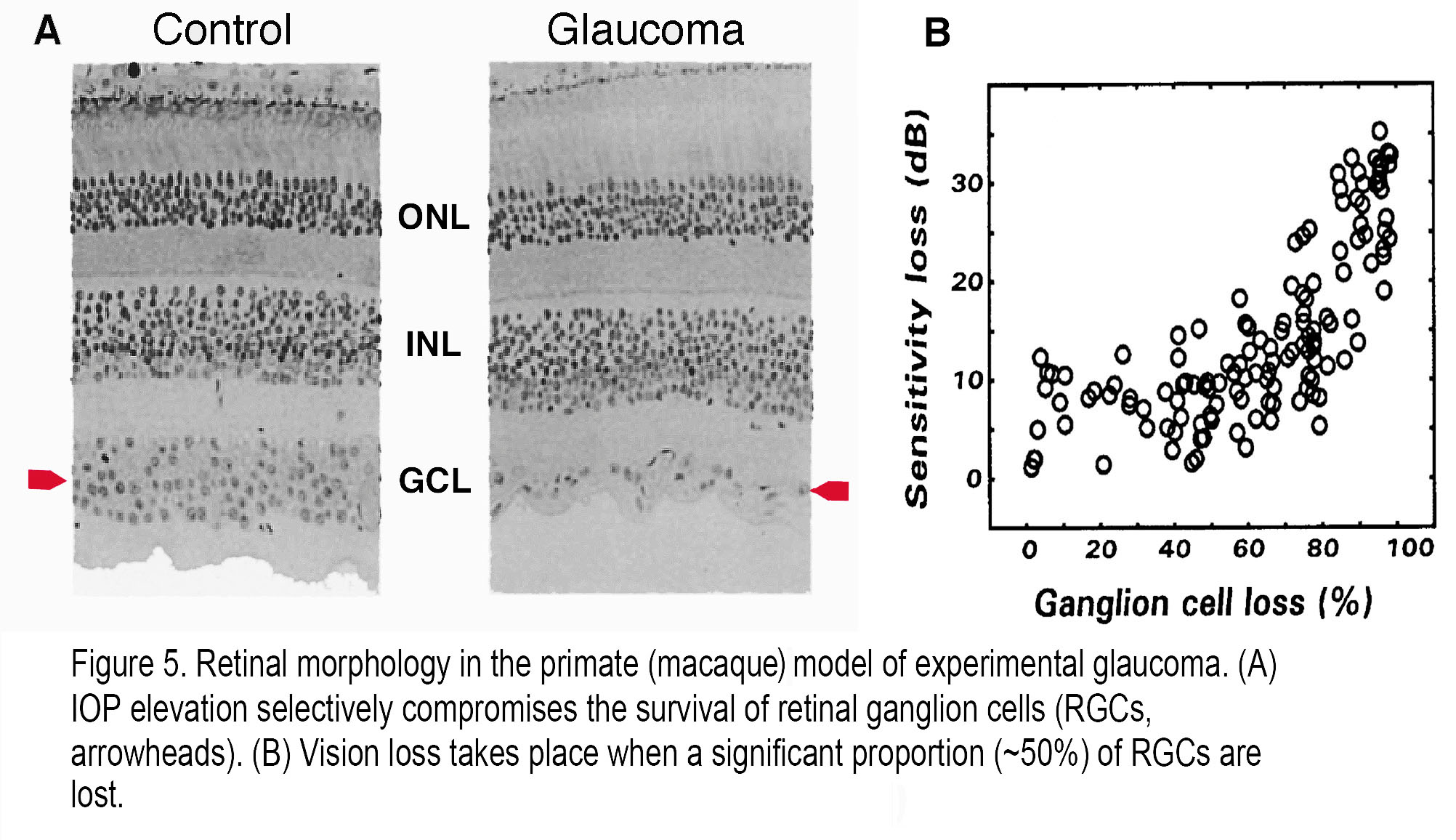 图5。实验性青光眼灵长类(猕猴)模型的视网膜形态。(A)眼压升高选择性地损害了视网膜神经节细胞(RGCs,箭头)的存活。(B)当很大比例(~50%)的rgc丢失时,就会发生视力丧失。
图5。实验性青光眼灵长类(猕猴)模型的视网膜形态。(A)眼压升高选择性地损害了视网膜神经节细胞(RGCs,箭头)的存活。(B)当很大比例(~50%)的rgc丢失时,就会发生视力丧失。
青光眼患者经常描述经历彩色“光晕”,这可能反映了角膜上皮水肿和ONH的短暂压缩。许多传闻病例将强烈的情绪创伤与疾病的急性表现联系起来,但情绪/焦虑状态与青光眼之间的关系仍有待系统评估。与对非人类灵长类动物的丰富研究相比,对青光眼人类视网膜的组织学研究相对较少。Pavlidis及其同事在2003年的一项研究中展示了一个例子(图6)(9)。与猕猴一样(图5),晚期青光眼患者的视网膜中几乎没有神经节细胞保留。与正常的小神经节细胞(图6,图K)相比,染色的罕见神经节细胞萎缩,很少有树突状分支,失去浓密(图6,图K),细胞体萎缩,神经纤维层中明显有珠状轴突(图6,图B,C,E, F和H, I)。图A, D, G和J)。更大的细胞类型,如阳伞神经节细胞,在晚期青光眼视网膜中同样受到影响(9).有关人类视网膜神经节细胞类型,请参阅“beplay13官网 .对啮齿动物模型,见本章后面,并由Nickells等人(2012)(10)审查。
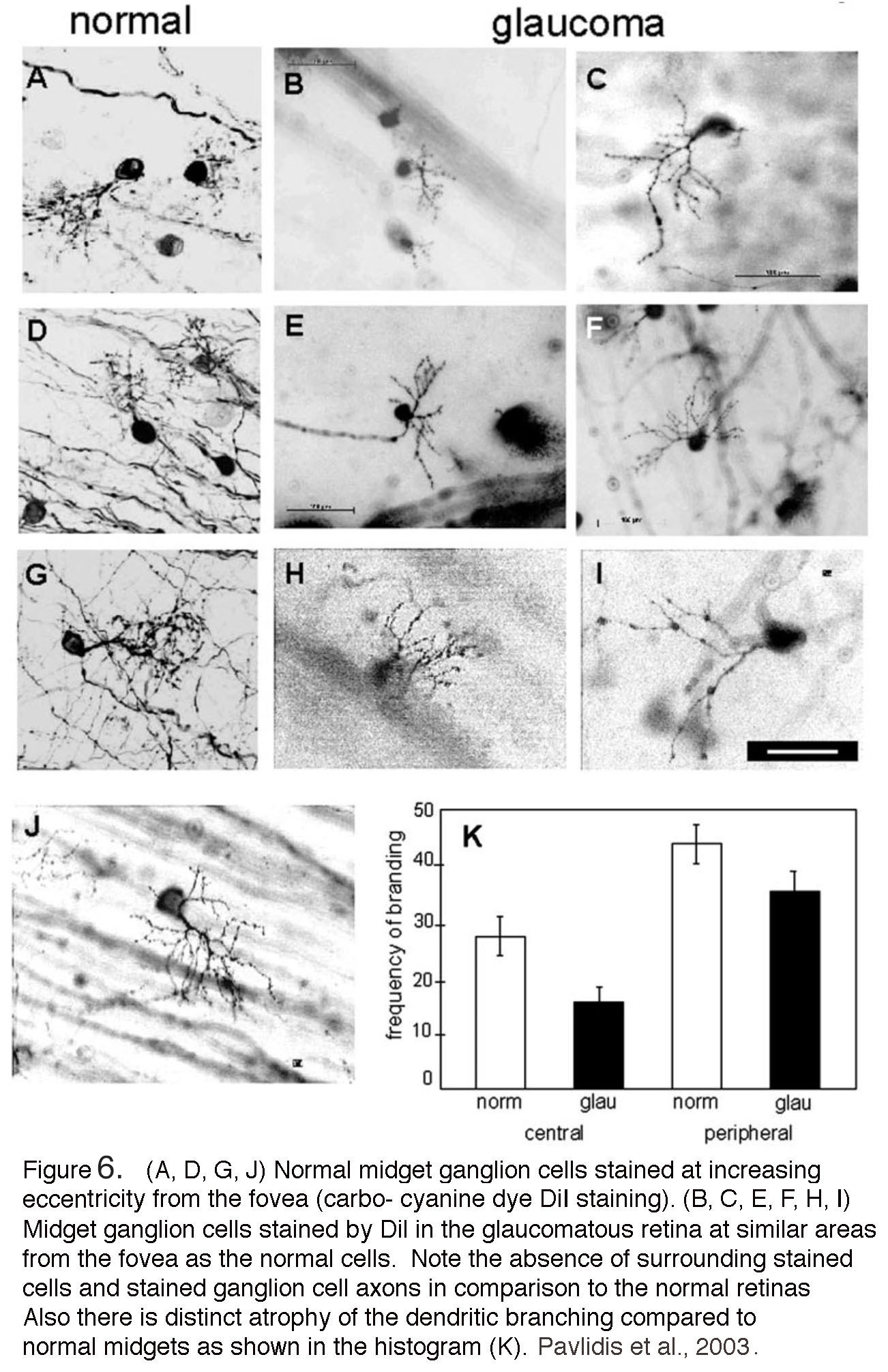 图6。(A, D, G, J)正常小神经节细胞在离中心凹越来越偏心处染色。碳菁染料(DiI)染色。(B, C, E, F, H, I)青光眼视网膜中DiI染色的小神经节细胞与正常细胞距离中央凹相似。与正常视网膜相比,周围无染色细胞和染色神经节细胞轴突。此外,与正常侏儒相比,树突分支有明显的萎缩,如直方图(K)所示(来自Pavilidis et al, 2003)。
图6。(A, D, G, J)正常小神经节细胞在离中心凹越来越偏心处染色。碳菁染料(DiI)染色。(B, C, E, F, H, I)青光眼视网膜中DiI染色的小神经节细胞与正常细胞距离中央凹相似。与正常视网膜相比,周围无染色细胞和染色神经节细胞轴突。此外,与正常侏儒相比,树突分支有明显的萎缩,如直方图(K)所示(来自Pavilidis et al, 2003)。
类型的青光眼
“原发性”青光眼表现为眼压升高或不升高的视神经病变(即,疾病的原因尚不清楚),而“继发性”青光眼则由眼压升高高于正常的已知病理机制定义。这两种疾病都有共同的前眼功能障碍、症状(RGC丧失和视神经病变)和治疗(眼压降低)。诊断仍然是一个问题,因为青光眼往往是在许多rgc(图1中的绿色细胞)不可挽回地丢失后才被诊断出来的。青光眼最常见的形式没有孟德尔遗传,但基因和种族背景显然很重要,因为不同民族青光眼的流行类型有显著差异。例如,原发性开角型青光眼(POAG)在非洲很普遍(4.2%),而原发性闭角型青光眼(PACG)在亚洲更常见(1.1%)。然而,就人口规模而言,亚洲人仍然占全球POAG病例的大多数(~53%)(2,11)。一般来说,具有较大生物学效应的基因突变(如视神经病变)是罕见的,而与较小生物学效应相关的基因变异则很常见(12)(见下文)。对所有类型青光眼的治疗仅限于药物降低眼压(唯一可治疗的危险因素),在极少数情况下,前眼手术和激光光/热凝治疗,但对神经退行性变没有治疗方法(见(13-15))。
原发性开角型青光眼
POAG是最常见的青光眼形式,在西半球占青光眼病例的80%。“角度”指虹膜和角膜之间的虹膜角膜裂口,在正常眼和POAG中是“开放的”(图7A和图7B),为房水流入小梁网和Schlemm管提供通道。临床上,POAG的诊断标准为开角、眼压> 21 mm Hg和视野丧失与神经节细胞轴突退行性变相关(“视神经病变”)(16)。虽然在50岁以下相对少见,但80岁以上的美国人中有近8%患有POAG;非裔美国人的发病率是白种人的5倍,非洲裔美国人发病更早,病程进展更快(17)。另一方面,在评估每十年POAG的优势比(18)时,欧洲血统的人比非洲人或亚洲人更容易受到伤害。男性的患病率似乎更高。在大多数POAG病例中,患者可能发展到完全失明而不经历任何疼痛或不适,但当张力率非常高,眼压超过~50 mm Hg时,也可能发生疼痛或不适。
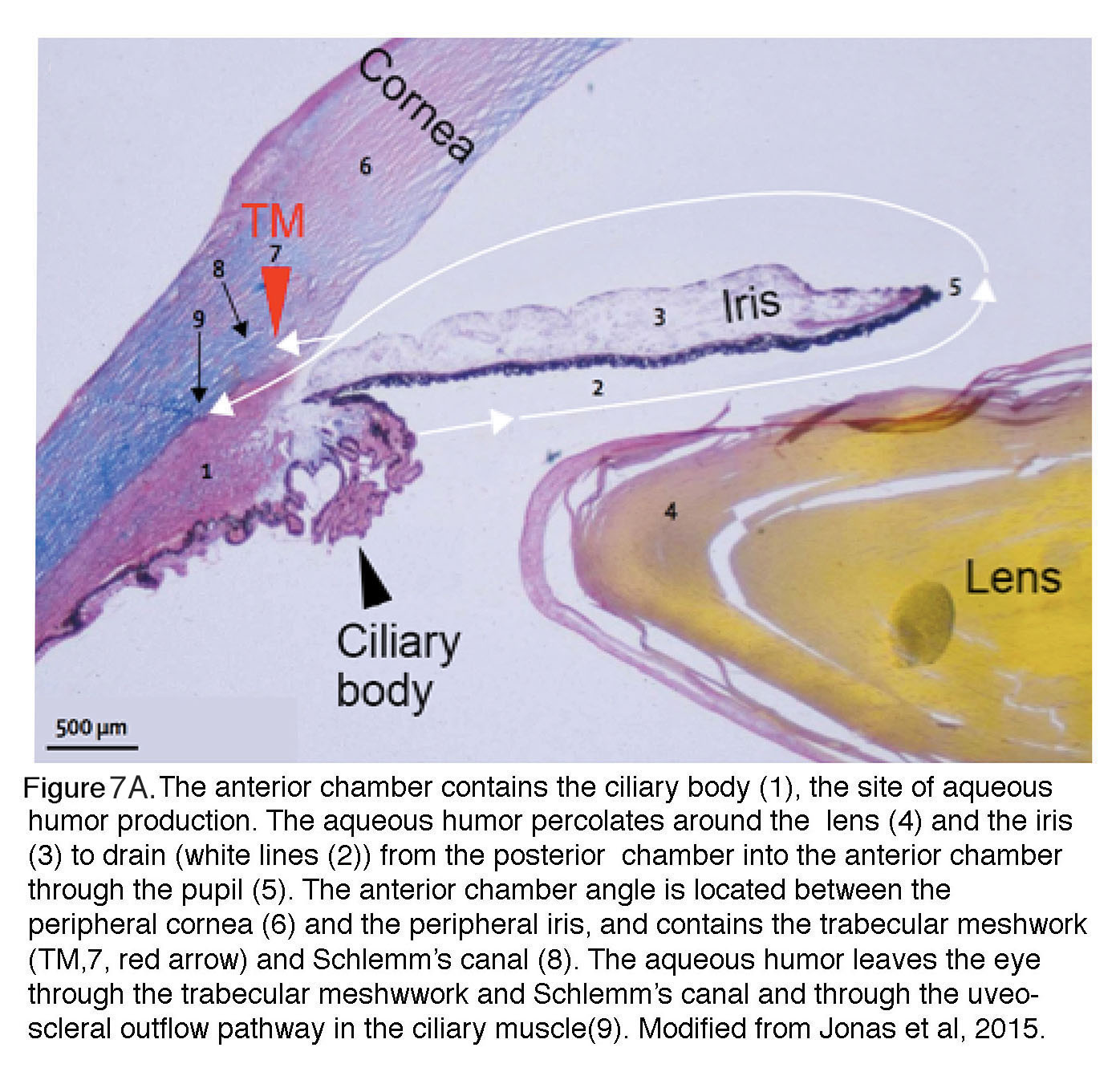 图7。前房包含睫状体1即房水产生的部位。房水在晶状体周围渗出4和虹膜3.排干(白线2)从后房经瞳孔进入前房5.前房角位于周围角膜之间6和虹膜周围,它包含小梁网(TM7红色箭头)和施莱姆运河8.房水通过小梁网和施莱姆管,以及睫状肌中的葡萄膜-巩膜流出道离开眼睛9.修正自Jonas等,2017(13)。
图7。前房包含睫状体1即房水产生的部位。房水在晶状体周围渗出4和虹膜3.排干(白线2)从后房经瞳孔进入前房5.前房角位于周围角膜之间6和虹膜周围,它包含小梁网(TM7红色箭头)和施莱姆运河8.房水通过小梁网和施莱姆管,以及睫状肌中的葡萄膜-巩膜流出道离开眼睛9.修正自Jonas等,2017(13)。
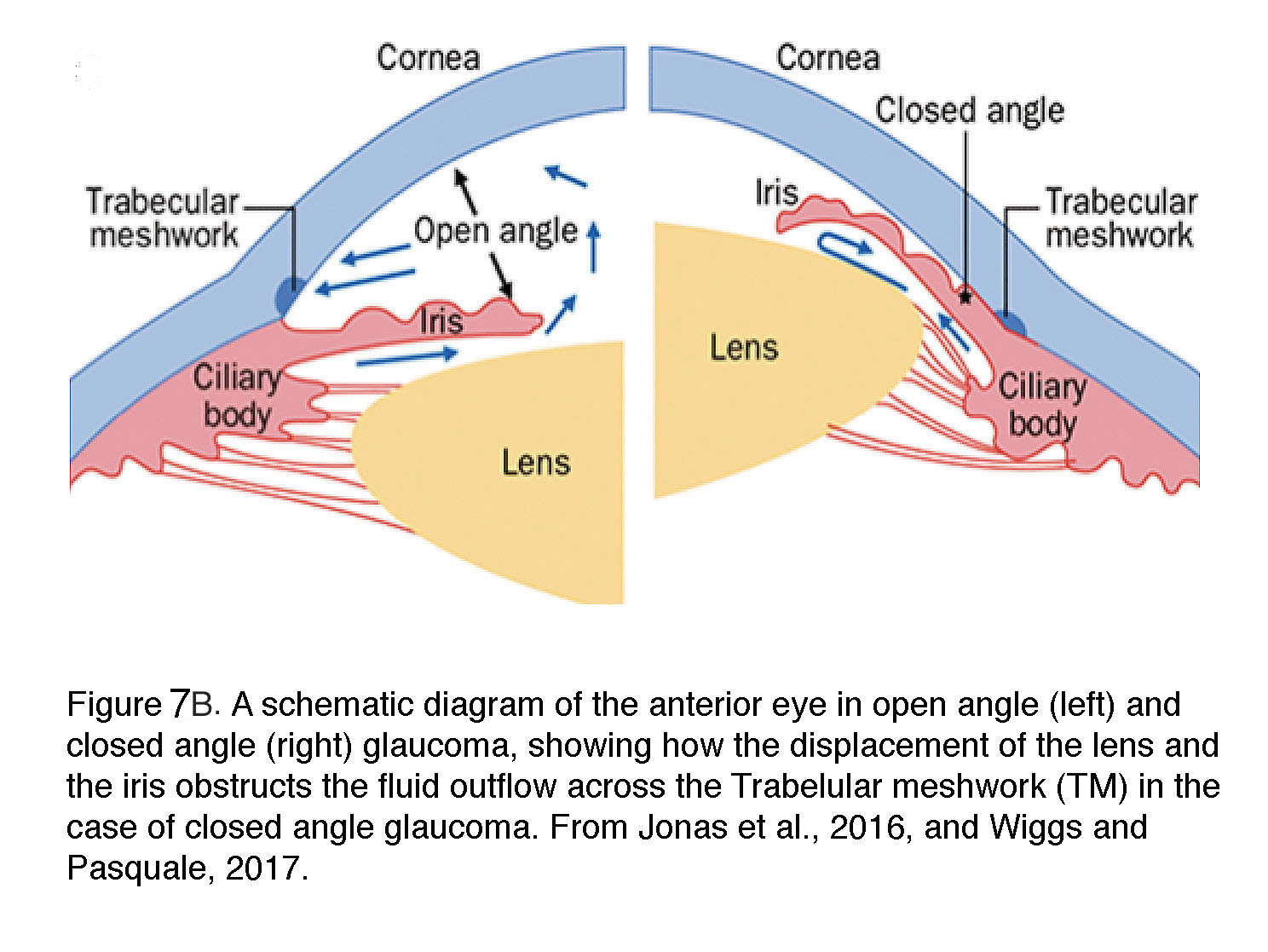 图7 b。开角型青光眼(左)和闭角型青光眼(右)的前眼示意图,显示闭角型青光眼时晶状体和虹膜的移位如何阻碍液体通过小梁网(TM)流出。来自Wiggs和Pasquale, 2017(12)。
图7 b。开角型青光眼(左)和闭角型青光眼(右)的前眼示意图,显示闭角型青光眼时晶状体和虹膜的移位如何阻碍液体通过小梁网(TM)流出。来自Wiggs和Pasquale, 2017(12)。
一个关键的遗传性OAG风险因素是眼压,其变化范围在~7 - > - 20mmhg之间。在任何5 - 9年期间,基线每增加1 mmhg,发病风险就会增加10-14%(19)。眼压在21 - 25mm Hg范围内的未治疗眼从早期到晚期POAG进展约14.4年,但眼压在> ~ 30mm Hg时滞后期缩短至约2.9年。近视个体存在眼压升高和POAG升高的风险,可能是由于结缔组织支持较弱(18)。值得注意的是,这种疾病是不可预测的,因为有相当一部分眼压升高的患者没有表现出轴突变性、视野缺陷和视力丧失的迹象。
POAG有明确的遗传基础,在双胞胎中遗传率约为0.95,患病个体的兄弟姐妹患该病的风险增加近8倍。与黄斑变性(AMD)相比,全基因组研究(GWAS)确定了约60%的负责基因,已知POAG序列只占已知病例的不到10%。其中包括约50个基因位点,三个孟德尔基因——心肌素(MYOC)、视神经素(OPTN)和TANK结合激酶(TBK1)的突变占POAG病例的约5%,而其他基因或环境因素的影响很小(12,20)。MYOC是一种功能未知的钙结合蛋白,在TM细胞、视网膜色素上皮和星形胶质细胞中表达。缺乏或过表达MYOC的小鼠和人类的房水引流正常,不会产生高眼压,而表达人类嗅觉素域Tyr437His突变的小鼠的眼压升高和视神经损伤,反映了人类青光眼(21)。约90%的MYOC突变携带者患青光眼,可能是由于蛋白质-蛋白质相互作用紊乱、细胞内错误折叠蛋白质积累和ER/内体应激(12)。OPTN编码一个支架蛋白,它至少有23个结合伙伴,8个蛋白质结合位点,并可能在多种信号通路中起作用,包括TM细胞和rgc中的自噬和囊泡运输(22)。
POAG的普遍观点是一种多因子疾病,其中多个基因、蛋白质和脂质信号通路失调,但也可能受到环境应激源和表观遗传机制的影响。支持POAG的压敏和不敏方面的分子“相互作用组”仍有待完全描述,其潜在线索来自最近的全基因组关联研究(GWAS)对成人发病POAG基因座的分析,包括编码胆固醇结合小洞穴蛋白CAV1/2、ABCA1 (atp结合盒转运体1)、TGFBR3(转化生长因子β受体3)、FOXC1(叉头盒C1)、ELOVL5(长链脂肪酸家族成员5的延伸)、ATOH7(无调性同源物7),CYP1B1(细胞色素P450酶)和ATXN2(共济失调蛋白亚型2)蛋白。就其本身而言,这些蛋白可能是无害的-和/或独立于IOP -但当与病理机械转导结合时可能积累风险。独立于op的基因[例如,SIX6(一种转录因子)和CDKN2AS(一种非编码RNA)](12)可以复合压力介导的损伤。一些GWAS位点可能是种族特异性的:脂质筏-调节细胞机械转导的大分子复合物-在欧洲队列中通过CAV1和CAV2的突变与POAG相关(23)。因此,降胆固醇药物(如他汀类药物)可能会降低这些人群患POAG的风险(24)。除了基因组成的改变,青光眼的风险也可能与拷贝数的变化(碱基缺失/重复)有关(25)。
前眼信号机制的压力依赖性失调反映在房水成分中,它显示了大量信号分子水平的改变,包括腺苷、一氧化氮、ATP、内皮素-1 (ET-1)、脂质(内源性大麻素、前列腺素、胆固醇)、转化生长因子(TGFβ)、CTGF(结缔组织生长因子)、血管紧张素II和耳蜗素。ET-1和TGFβ2调节小梁通路内ECM分子的沉积,进而可能影响房水流出(26)。另一种感兴趣的蛋白质是CYP1B1,它是细胞色素P450酶联盟的成员,负责代谢花生四烯酸下游的脂肪酸分子。CYP1B1突变与儿童和年轻人的青光眼相关(2),该蛋白在青光眼相关的眼组织中表达,如小梁网、视网膜和睫状体,在这些组织中它可以影响细胞对机械应激源的敏感性(27)。钙透性TRPM3通道(在睫状体和晶状体中表达)是高张力青光眼和白内障的联合危险因素(28)。其他重要的信号机制包括Rho和MAP激酶,它们调节蛋白水解酶、促炎细胞因子、自噬和收缩力的表达(29,30)。
正常眼压青光眼(NTG)
约30%的POAG患者出现NTG,其中变性rgc在“正常”IOP时表现为典型的轴突损失和视野缺陷沙漏模式(31),即水平始终低于21 mm Hg。危险因素包括女性、种族(日本人比白种人更常见)、血管失调和低血压(32)。两个患病率为1-2%的NTG基因(OPTN和TBK1)与POAG共享,并编码与自噬(细胞内蛋白质降解机制)相关的蛋白质(33)。通过视神经头的异常低脑脊液压力引起的压力梯度可增加NTG的轴索损伤(32)。降低眼压方案改善了一些患者的预后:6年后,约55%接受眼压降低药物治疗的患者视力稳定,而未接受治疗的患者视力稳定的比例为约40%(31,34)。考虑到NTG患者的病理反应似乎涉及无害的机械压力源,这些患者可能具有功能获得突变,增加了他们对IOP的敏感性。类似的对机械压力源的超敏(痛觉过敏)是神经性疼痛机制的一个众所周知的特性,涉及周围神经系统中机械敏感离子通道的功能改变(35)。
原发性闭角型青光眼
闭角型青光眼在东亚人群中很常见,在白种人中很罕见,是造成严重视力丧失患者不成比例的原因。该病是由虹膜、晶状体和晶状体后结构的紊乱引起的,它们通过缩小虹膜和角膜之间的角度来阻碍房水的引流(图7B)。它更可能发生在妇女和高度远视的人身上。在大约三分之一的PACG患者中,眼压突然升高(通常超过30毫米汞柱)会引发急性症状(剧烈疼痛和恶心、结膜充血、角膜水肿和呕吐),需要立即进行医疗干预(12)。该病具有高度遗传性,基因相关性研究涉及编码细胞外基质(ECM)分子(如MMP、HSP70、NOS3)或眼部发育(卷曲相关MFRP蛋白、肝细胞生长因子HGF)多态性的基因。蛋白糖基化(DPM2)、乙酰胆碱生物合成(CHAT)、ST18(抑制致瘤性18)和转录调节(GLIS3)(36)。毫不奇怪,许多这些风险基因编码与细胞粘附、ECM内稳态和机械转导(局部粘附、细胞连接、胶原蛋白)相关的过程。
急性PACG后可能会有一段时间眼压正常,需要几天或几周时间从休克中恢复,并开始以正常速度产生房水,使眼压升高到符合闭角程度的水平(12)。
继发性青光眼
继发性青光眼反映了由于病理性产生和/或房水引流减少导致眼压升高至正常范围以上的病理机制。变型包括假性剥脱性青光眼、新生血管性青光眼、色素性青光眼和激素性青光眼。Pseudoexfoliative青光眼当蛋白质团块聚集在前房阻塞通过小梁网的流出时发生。它在斯堪的纳维亚人群中发病率较高,与帕金森病和阿尔茨海默病的风险因素相同,并与赖氨酸氧化酶样1 (LOXL1)基因突变以及编码CACNA1A、TMEM136、SEMA6A和其他基因的位点突变有关。新生血管性青光眼:虹膜角膜角(“虹膜簇”)的血管增生导致纤维物质泄漏,从而闭合角膜角。与视网膜缺血有关。危险因素包括糖尿病。类固醇青光眼发生于服用类固醇药物抑制黄斑水肿、角膜移植排斥、免疫高反应性和损伤相关炎症的患者。由此导致的小梁网结构变化(包括MYOC的诱导)增加了其对房水流出的阻力并提高了IOP。如果不治疗,这将导致40%的患者(“类固醇反应者”)发生POAG(37)。颜料的青光眼通常源于虹膜内色素上皮细胞萎缩引起的“色素分散综合征”。黑色素扩散到小梁网,阻碍了房水流出,提高了眼压。DBA/2J小鼠品系,发展出类似的表型慢性青光眼,有突变Gpnmb基因(编码溶酶体相关的糖化蛋白)和Tyrp1基因(编码一种可能具有酶和结构功能的黑素体蛋白)(38,39)。其他类型的青光眼与白内障形成、某些眼部肿瘤、葡萄膜炎(眼部炎症)、近视和早发性青光眼有关。
衰老和青光眼
衰老和细胞衰老是癌症的主要危险因素进展青光眼。我们每年损失10,000个神经节细胞(0.3 - 0.6%)和0.58%的TM细胞——到80岁时,一个眼压正常的健康个体失去约30%的RGCs(8)。老年人神经节细胞的损失与老年人认知、运动和其他功能的普遍下降有关,与青光眼有许多相似之处,包括树突重塑、视神经轴突运输减少(在啮齿动物的一生中减少一半),神经节和小梁细胞质量控制机制降低,损伤后恢复率降低。97.6%的40岁以上患者诊断青光眼的中位年龄为64岁,患病率从50 - 59岁的0.2% - 2.7%急剧上升到80岁(40岁)后的1.6% - 12.8%。
虽然年龄是POAG的一个强有力的预测因素,但我们不知道是什么临界压力传递给老年人的rgc,使他们对眼压升高的抵抗力降低。一个可能的原因可能是眼结构的硬度增加(“眼硬度”),它可能增加小梁收缩力(提高眼压),增加神经节细胞和胶质细胞对压力介导的应激的敏感性。老年人还可能有眼部代谢缺陷,并伴有年龄依赖性氧化应激、线粒体功能障碍(41)和神经保护机制丧失(42)。
眼内压的产生
注意到这一点很重要IOP反映了房水产生和流出之间的平衡(图7A和图7B)。Accott等人(2014)(43)和Braunger等人(2015)(26)在评论中讨论了房水调节的细节,但需要注意的是,这种透明的、有营养的液体是由纤毛体持续产生的(健康成人在白天约2.4 μ l/min;图7A中的黑色箭头)以提供无血管化晶状体。房水通过小梁网(TM;红色箭头,图7A中的#8)。在灵长类动物中,TM支持约80-90%的房水流出,因此是液体流出的主要瓶颈,而睫状肌中的辅助“葡萄膜”流出通道(图6A中#1)支持其余部分。夜间的房水流出量比白天低约50%,因此在睡眠中发生急性压力诱发眼损伤的风险增加(44)。含水液体的异常产生或通过TM和Schlemm管(一种由内皮样细胞组成的管,将从TM收集的液体输送到静脉系统)引流不足都可能导致青光眼。
眼压依赖性在青光眼病例中占很大比例。任何水平的眼压都可能导致病变,但当慢性眼压超过20 mm Hg时,患青光眼的风险会呈指数级增加(图8)。眼压的大小和持续时间与疾病的严重程度相关,较大的眼压升高会加速疾病的发生和进展(AGIS 2000)。同样,眼高压持续时间越长,发生视神经病变的可能性越高(31)。重要的是,许多眼压升高的个体不会发展为青光眼,多达50%的青光眼患者(“正常张力”患者)可能没有统计上的眼压升高(45)。这些患者可能表达的保护因子抵消了导致神经退行性变的致病因子,而“正常张力青光眼”患者可能表达的功能获得换能器机制使他们对机械压力超级敏感。
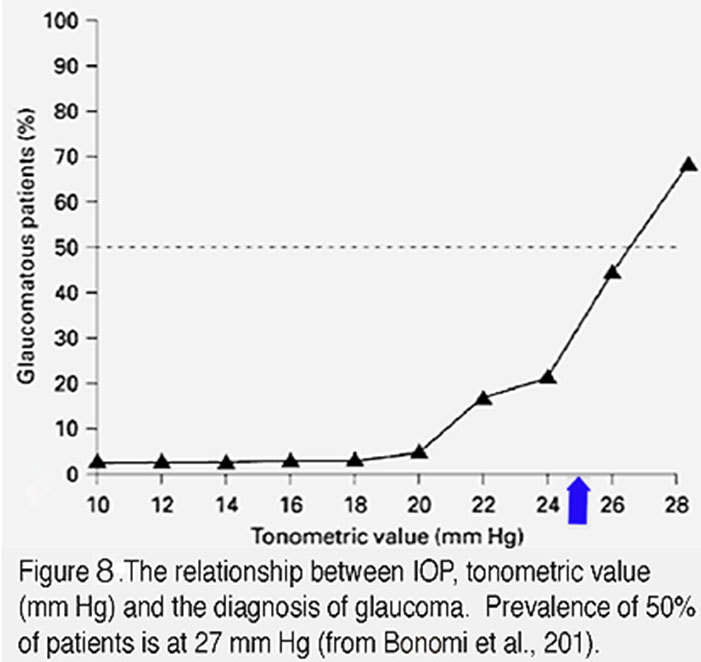 图8。眼压、眼压计值(mmhg)与青光眼诊断的关系。50%的患者患病率在27 mm Hg。来自Bonomi等,2001(138)。
图8。眼压、眼压计值(mmhg)与青光眼诊断的关系。50%的患者患病率在27 mm Hg。来自Bonomi等,2001(138)。
眼压升高是青光眼的危险因素之一
脊椎动物的眼睛是一个高度动态的环境,眼压提供了维持眼组织形状、生长和光学特性所必需的静水压力。随着年龄的增长,眼压升高(IOP)是青光眼发生和发展的最重要的危险因素(图8)。眼压依赖性约占青光眼病例的30 - 70%(46)。新生啮齿动物和灵长类动物的平均眼压在6 - 8毫米汞柱之间,成年后增加到7 - 15毫米汞柱。右眼的眼压振幅往往略高于左眼,但个体眼的眼压振幅的终生变异性非常小(<2毫米汞柱)(31)。与整个生命周期中保持稳定的平均眼压振幅相比,眼压在时域上是复杂的,表现为搏动、昼夜周期和振荡(~ 3mmhg ~ 1hz)(47)。眨眼、抓挠眼睛、打喷嚏、调节晶状体、动脉血压和眼球运动都会使眼压瞬间升高(最高可达200毫米汞柱)。眼压波动的幅度和节律受Cry1/Cry2时钟基因和褪黑激素局部释放的调节,褪黑激素在夜间达到峰值,在青光眼患者中更为明显(48)。体位(如瑜伽倒置练习)、举重和泳镜可能会增加眼压和青光眼的风险,而适度的体育锻炼和富含ω3/ω6多不饱和脂肪酸的饮食可使眼压降低约4 - 5毫米汞柱。鉴于各种各样的危险因素和青光眼相关的信号传导机制,没有明确的眼压阈值使我们能够始终确定视神经病变的风险患者。
目前接受的青光眼治疗很大程度上局限于降低眼压,如果眼压能降低约30-50%,这种策略通常可以阻止疾病的发展。抗青光眼药物针对睫状体和/或继发(葡萄膜巩膜)流出通路中的水基液体产生。因为降低眼压也可以减少“正常”眼压(8-15 mm Hg)水平的患者视力下降的进展(17),青光眼可以被视为在任何眼压水平下,眼睛对机械压力的易感性增加的疾病.不幸的是,在相当一部分(25-50%)患者中,最常用的IOP降低剂拉托前列素(前列腺素e2 α类似物)并不能将IOP降低20%以上,而高达6%的POAG患者无反应或不耐受任何IOP药物(31,45)。一些在实验范例中显示出疗效的治疗方法,如靶向CB1受体(49)的大麻素、血管紧张素II受体阻滞剂、褪黑素受体3、腺苷受体1激动剂、TRPV4通道、TREK-1通道和内皮素受体1阻滞剂,尚未完成临床试验。直到2017年,才有药物能够靶向主要(小梁)流出机制,当时几种Rho激酶(ROCK)抑制剂(如利帕舒地尔)被批准用于POAG的局部治疗。不幸的是,令人不快的副作用(如充血)可能限制了它们对许多患者的作用。因此,开发靶向小梁流出成分而无副作用的药物应该是优先考虑的问题。
小梁网(TM)的细节
TM细胞通过控制液体流入Schlemm管,在IOP调节中发挥核心作用(图7A)。这些细胞形成三个相邻的层(葡萄膜层、角膜巩膜层和近巩膜层),它们在基因表达模式和对吞噬作用、液体过滤、硬度和液体流动阻力的相对贡献(50)方面不同,但很难从个体发育、形态或功能的角度进行分类:(i)与巨噬细胞相似,它们表达组织相容性和其他免疫标记,并显示吞噬细胞样清除活性;(ii)与胶质细胞相似,具有重要的体积调节功能,参与炎性细胞因子(如IL-1α、TNFα)的释放;(iii)与内皮细胞相似,形成具有抗血栓和抗原递呈功能的屏障;(iv)与成纤维细胞相似,它们释放细胞外基质并促进收缩(v)它们表达平滑肌肌动蛋白(αSMA),一种典型的平滑肌细胞蛋白。肌动球蛋白介导的压力诱发的TM收缩被认为通过增加流出通道对液体流动的阻力来提高IOP(30)。流出阻力的慢性增加是眼压升高、细胞骨架硬化和收缩力增强之间的正反馈.换句话说,细胞骨架和ECM重塑是高血压青光眼细胞学和功能特征的基础.机械拉伸和病理之间类似的正反馈在其他组织中也有描述,包括肺和心脏,在这些组织中,细胞感知到组织硬度的微小变化会导致严重的纤维化重塑。
在亚细胞水平上,压力的增加诱导TM细胞中肌动蛋白应力纤维、粘附分子和细胞外基质(ECM)的重构上调(图9C和D),以及与细胞粘附、代谢、离子运输、炎症和细胞周期相关的数百个基因的上调/下调(29)。尽管这种动态响应的每一步都不清楚,但最近的研究表明,机械应力诱导TM细胞(图9A)和分别调节水基液体流出和流入的纤体细胞胞内钙浓度[Ca2+]i的增加(51)。钙反过来调节细胞生理的几乎每一个方面,包括调节收缩力、僵硬度和细胞因子TGF-β水平的增加,丝裂原激活蛋白(MAP)激酶的激活,金属蛋白酶(mmp)的释放和ECM分子的沉积,如纤维连接蛋白和versican(图9D)。压力对体外和体内小梁流出和IOP调节模型中[Ca2+]i和细胞骨架重塑的影响,通过抑制拉伸激活的TRPV4通道(图9B)在实验中被打破,这触发了立即的IOP降低(51)。
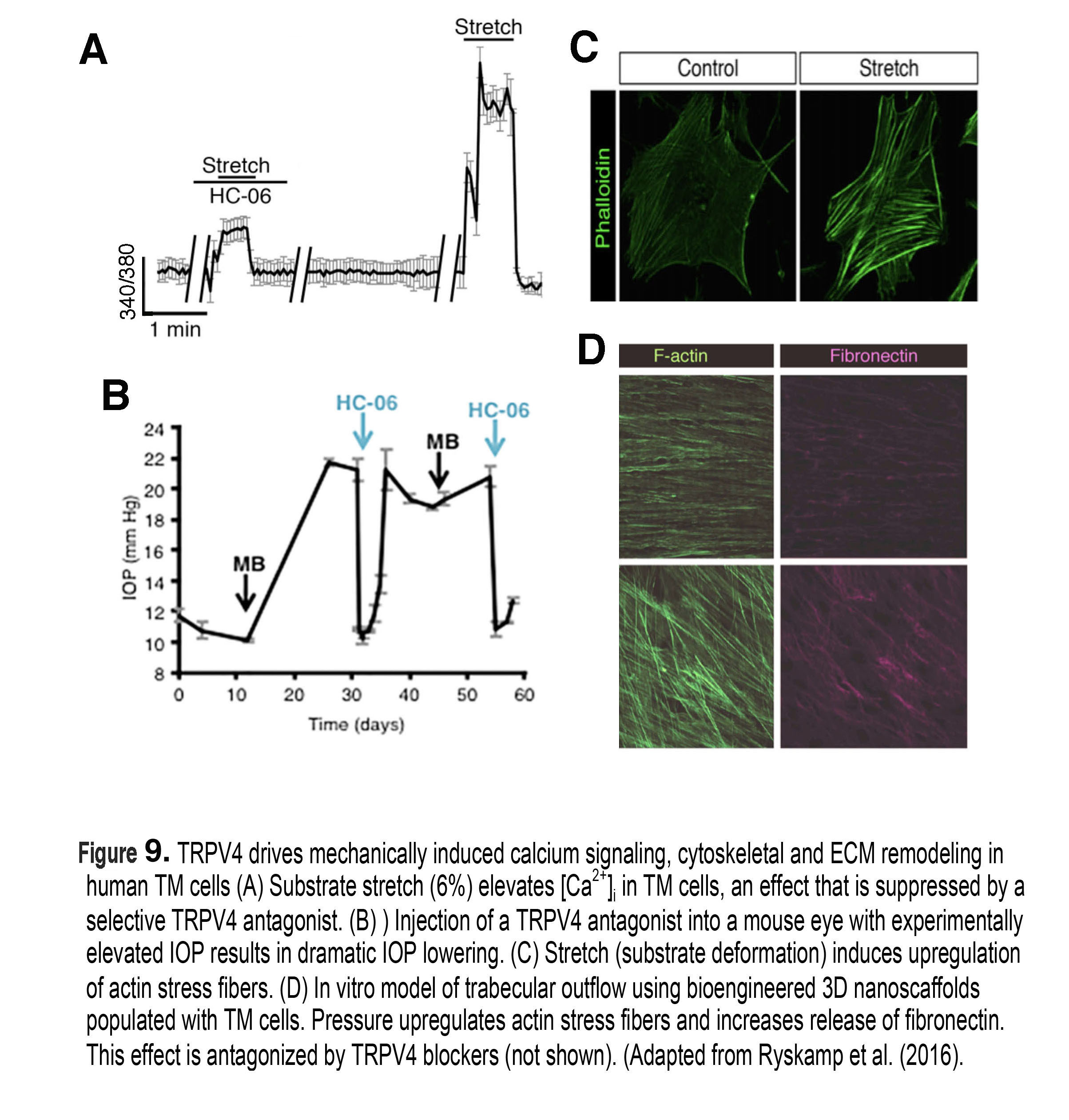 图9。TRPV4在人TM细胞中驱动机械诱导的钙信号、细胞骨架和ECM重构。(A)底物拉伸(底物变形,6%)会提高TM细胞中的[Ca2+)i,这一效应会被选择性TRPV4拮抗剂抑制。(B)将TRPV4拮抗剂注射到实验性眼压升高的小鼠眼睛中,可显著降低眼压。(C)拉伸驱动腹侧肌动蛋白应力纤维上调。(D)利用生物工程3D纳米支架植入TM细胞建立体外小梁流出模型。压力上调肌动蛋白应力纤维,增加纤维连接蛋白的释放。这种效应被TRPV4阻滞剂拮抗(未显示)。改编自Ryskamp等人(2016)(51)。
图9。TRPV4在人TM细胞中驱动机械诱导的钙信号、细胞骨架和ECM重构。(A)底物拉伸(底物变形,6%)会提高TM细胞中的[Ca2+)i,这一效应会被选择性TRPV4拮抗剂抑制。(B)将TRPV4拮抗剂注射到实验性眼压升高的小鼠眼睛中,可显著降低眼压。(C)拉伸驱动腹侧肌动蛋白应力纤维上调。(D)利用生物工程3D纳米支架植入TM细胞建立体外小梁流出模型。压力上调肌动蛋白应力纤维,增加纤维连接蛋白的释放。这种效应被TRPV4阻滞剂拮抗(未显示)。改编自Ryskamp等人(2016)(51)。
在动物和人类中,眼压的慢性增加会触发成千上万的小梁和视网膜基因的上调和下调,其中许多基因编码与神经胶质激活、补体基因上调、内皮功能障碍和RGC退化相关的信号通路。杰克逊实验室(Jackson Laboratories)的青光眼发现平台(Glaucoma Discovery Platform)是展示这些途径复杂性的一个有用的在线资源。http://glaucomadb.jax.org/glaucoma).
视网膜神经节细胞的细节
哺乳动物有>45类RGCs -投射神经元具有独特的输入、功能特性和对大脑的轴突投射(G,图10中的红色细胞)。动物模型的体外和体内研究(主要是非人类灵长类动物和小鼠)证实了rgc对许多类型的机械应力的不成比例的脆弱性,包括IOP、压缩、拉伸和长时间的肿胀(35,52,53)。rgc的压力诱导损伤的空间模式与它们在ONH中的轴突位置相关,而其他类别的视网膜神经元往往(至少在最初)不受op诱导的损伤(图5)(8,54)。由IOP诱导的分子和细胞变化(转录改变、细胞器功能、钙信号、神经递质摄取、突触组织和细胞结构)反过来损害了细胞的功能,表现在它们的光响应、突触输出和存活。弄清楚为什么以及如何IOP优先靶向rgc目前是青光眼研究的一个核心挑战.
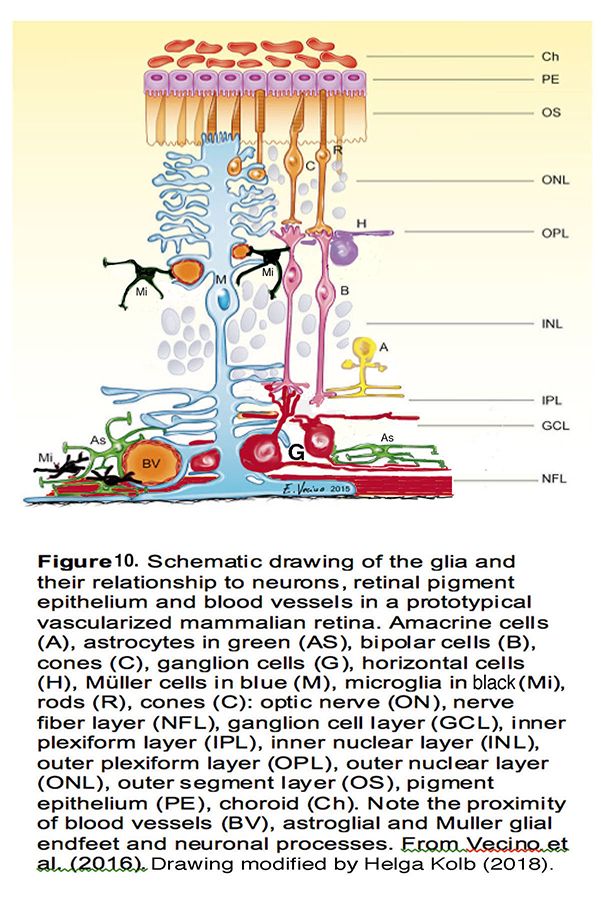
图10。在典型的血管化的哺乳动物视网膜中,胶质细胞及其与神经元、视网膜色素上皮细胞和血管的关系示意图。无分泌细胞(A)、绿色星形胶质细胞(AS)、双极细胞(B)、锥体细胞(C)、神经节细胞(G)、水平细胞(H)、蓝色Müller细胞(M)、黑色小胶质细胞(Mi)、杆状细胞(R)、视神经(ON)、神经纤维层(NFL)、神经节细胞层(GCL)、内丛状层(IPL)、内核层(INL)、外丛状层(OPL)、外核层(ONL)、外节段层(OS)、色素上皮(PE)、脉络膜(Ch)。注意靠近血管(BV),星形胶质细胞,Müller胶质端足和神经元突起。来自Vecino等人(2016)(139)。图由Helga Kolb修改。(2018)。
{kind=link}
虽然“青光眼”是视网膜损伤的晚期症状,但RGCs对压力的急性变化非常敏感,从而导致兴奋性立即改变、时空对比敏感度丧失和视力降低(55,56)。功能障碍与RGC树突收缩、变薄和复杂性降低、突触丢失和轴突运输变化相关(54,57 -60)(图11)。神经营养因子和细胞器的(顺行/逆行)运输受阻(61)加重了细胞放电和树突损伤的压力诱导变化,如果这种情况持续存在,就会导致视神经头的总体结构重构。ONH的宏观重塑往往在高眼压发生数月(啮齿动物)或数年(灵长类动物)后被观察到,时间滞后,此时进行神经保护干预往往为时已晚。相反,最近的研究表明,眼压诱导的树突结构改变和胶质激活(见下文)是青光眼的更好的早期标志,而不是运输损失。摩根的团队在一项具有里程碑意义的研究中强调了非轴索压力目标的重要性,该研究证明了这一点在完整的轴突存在的情况下,压力诱导的树突回缩和RGC丢失发生(62)。因此,早期压敏和非轴突靶点的研究(55,56)强调了早期神经保护治疗的必要性,其目标是保存视网膜内的树突形态和维持突触连通性。
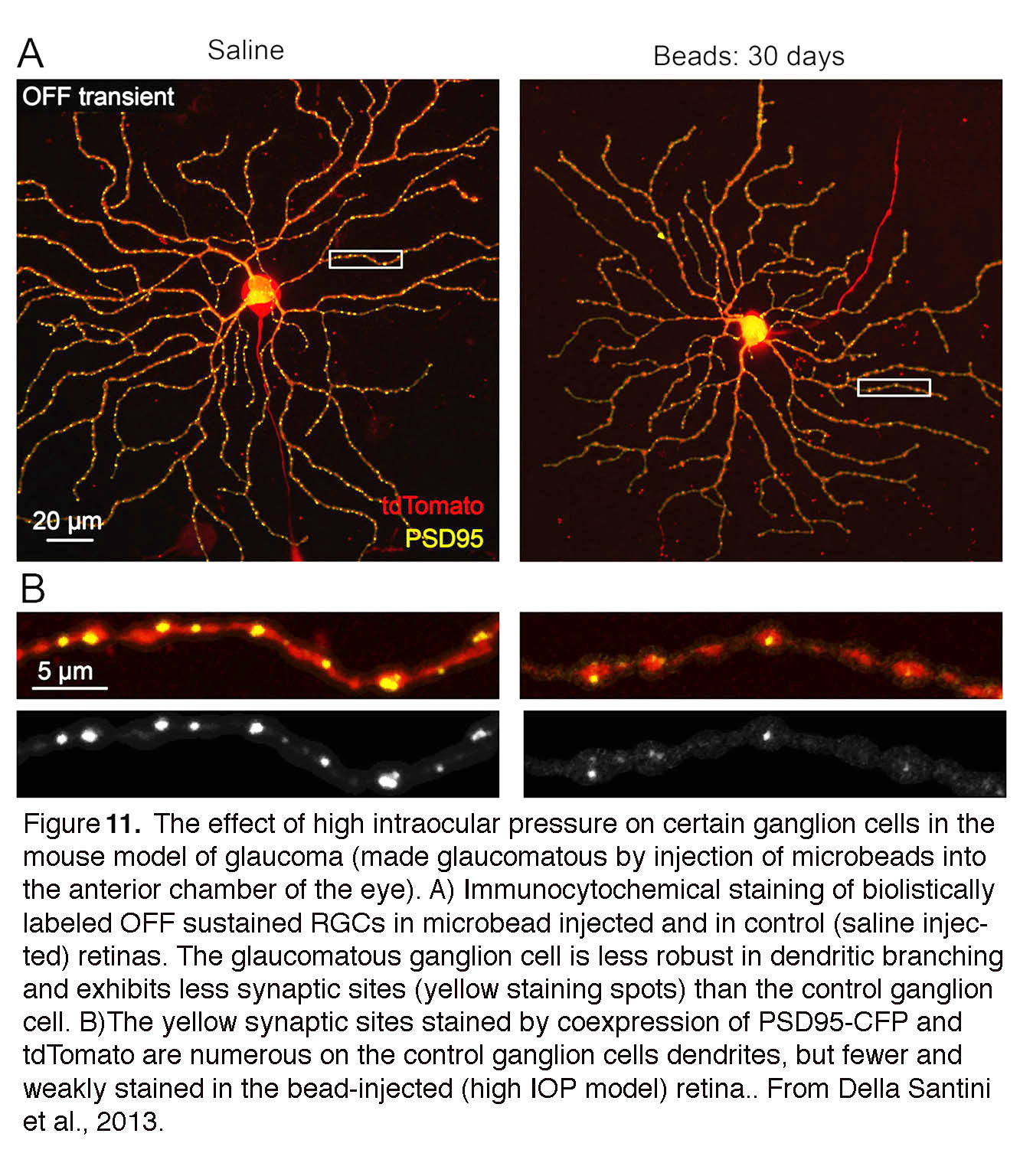 图11。高眼压对青光眼(前房注射微珠致青光眼)小鼠模型中某些神经节细胞的影响。(A)注射微珠和对照(盐水注射)视网膜中生物标记OFF持续视网膜神经节细胞的免疫细胞化学染色。与对照组相比,青光眼神经节细胞在树突分枝方面较弱,显示出较少的突触位点(黄色染色点)。(B)树突用tdTomato染色,突触部位用PSD95-CFP染色。对照组神经节细胞树突上的突触位点较多,而珠注组(高眼压模型)视网膜上的突触位点较少且染色较弱。摘自Della Santini等人,2013(55)。
图11。高眼压对青光眼(前房注射微珠致青光眼)小鼠模型中某些神经节细胞的影响。(A)注射微珠和对照(盐水注射)视网膜中生物标记OFF持续视网膜神经节细胞的免疫细胞化学染色。与对照组相比,青光眼神经节细胞在树突分枝方面较弱,显示出较少的突触位点(黄色染色点)。(B)树突用tdTomato染色,突触部位用PSD95-CFP染色。对照组神经节细胞树突上的突触位点较多,而珠注组(高眼压模型)视网膜上的突触位点较少且染色较弱。摘自Della Santini等人,2013(55)。
对于RGC亚型的压力诱发视神经损伤的发病和压力敏感性存在分歧。一项早期的灵长类动物研究报告称,周围核较大的RGC对压力异常敏感(63),但这些结果没有在灵长类动物(64)或啮齿动物中重现,这些动物的RGC损伤似乎是地形性的,而不是亚类特异性的(65)。iopo诱导的RGC丢失并不局限于视网膜的特定位置(55),核周大小和神经退行性变之间似乎没有什么相关性(58)。
为了更好地了解这种疾病,了解特定类别和子类型的rgc是否异常敏感或抵抗机械应力将非常有帮助。一些电生理记录结合报告器标记rgc的分析表明,OFF rgc对眼压升高比ON rgc更敏感(55,58),但另一项采用类似方法的研究得出了相反的结论(66)。同样,Ou等人(54)发现,在OFF细胞中,iopo诱导的树突重塑更为广泛,但其他研究显示,压力诱导的树突修剪在不同功能亚型之间没有明显差异(67)。这些差异可能是由不同的诱发高眼压的方式引起的。无论如何,青光眼早期RGC压损伤很可能反映分子/功能参数,而不是解剖参数(68)。例如,off型细胞的敏感性可能反映了来自局部血管或胶质细胞的不同反应,和/或营养因子可用性的改变(58)。本质上光敏的RGCs (iprgc)倾向于抵抗IOP升高(69)(参见Webvision关于黑视素表达神经节细胞的章节),可能是由于压力转导分子数量的减少(70)。
John的室室模型(39)超越了这种非此即彼的二分法,为眼压升高的早期影响提供了一个启发式框架。该模型通过假定青光眼中的RGCs变性涉及压力对树突、体突和轴突中分子不同的局部自毁程序的平行影响,从而深刻地推进了概念疾病范式。压力诱导损伤的一个合理的多室室调节因子可能是钙离子,它通过Ca2+依赖性蛋白酶、caspases、MAP激酶、内质网应激、自噬和凋亡级联来驱动树突、体细胞和轴突的重构(71-73)(见下文)。异常的自噬和内质网(ER)损伤,由于细胞内错误折叠蛋白的积累,钙稳态的丧失和蛋白质运输的不足,从而损害RGC的活力(73),代表了压力相关RGC应激的额外早期特征,而轴突收缩构成了可能的早期损伤和潜在的政变grâce,刺激细胞凋亡。与区隔化模型一致,局部轴突损伤可以通过减少氧化损伤,靶向双亮氨酸拉链激酶(DLK)酶和潜在的运动来改善。因此,旨在提高青光眼RGC生存率的临床前实验策略可以分为以下几种途径:(i)促进内源性保护机制的表达,(ii)向RGC提供外源性神经营养因子,(iii)靶向压力感知机制,(iv)防止下游重塑(v)通过靶向最终的共同凋亡通路减缓细胞死亡和/或(vi)使用广泛存在或作用机制尚不明确的神经保护药物。哺乳动物的成年神经节细胞在压力损伤后很少或没有再生反应,但通过重组和病毒表达的神经营养因子(如BDNF, CNTF, NT-4, FGF-2,神经turin)的传递,它们的存活时间可以延长很短的时间,这些神经营养因子在青光眼动物模型中部分保护RGCs(59,74)。在使用Rho激酶抑制剂、内皮素ET-2抑制剂、促红细胞生成素、白藜芦醇、NMDA受体拮抗剂、NO供体、抗氧化剂、α2-肾上腺素能激动剂(溴莫尼定)、大麻素(例如,Δ9-THC)和β-肾上腺素能阻滞剂治疗后观察到保护程度。美金刚胺是一种NMDA受体的拮抗剂,可以保护啮齿动物和非人灵长类动物的神经节细胞免受青光眼的伤害(75),但在人体试验中无效。 Enigmatically, exposure to long-wavelength red light (850 – 1100 nm) was reported to be protective (76), and visual (neuronal) activity was proposed to facilitate regeneration of injured RGC axons (77). The multiplicity and variability of protective effects suggests that many therapeutic strategies may have been targeting secondary molecular mechanisms that are downstream from the actual RGC mechanotransducers.
视神经头(ONH)。
ONH的退行性变(“拔罐”或“挖掘”)是目前将青光眼与其他RGC疾病(如非动脉性视神经前病变(NAION)、创伤性视神经病变、视神经炎和多发性硬化)区分开来的诊断标志。拔火罐与视觉测试(视野测量)中“弓形盲点”(敏感度丧失)的表现相关,与影像学研究中观察到的轴索损失相关。由于RGC丢失和视野缺陷的拓扑结构(人类为放射状,啮齿动物为扇形)与ONH内轴突的位置相关(10,38),ONH被广泛认为在决定驱动神经退行性变的最终轴突损伤程度方面很重要。
ONH包含RGC轴突的无髓鞘(早期)和有髓鞘(晚期)部分以及它们的细胞外基质ECM/胶质支架和毛细血管血液供应(图12和图13)(见(10,38))。该区域的生物力学受其周围巩膜的硬度和厚度以及ECM分子的胶质沉积的影响(78)。视神经进入视网膜的“乳头”(图12)是巩膜最薄弱的部分,因为轴突(人类约120万个)转90度,通过约200-400个大小不同的孔进入视神经,这些孔是称为视网膜结构的一部分板cribrosa.12%的轴突必须通过这些胶原毛孔。来自外周视网膜的轴突通过板的外周部分,而来自中央视网膜的轴突通过其中心,但来自同一视网膜偏心的轴突可能在视神经内的不同水平通过。
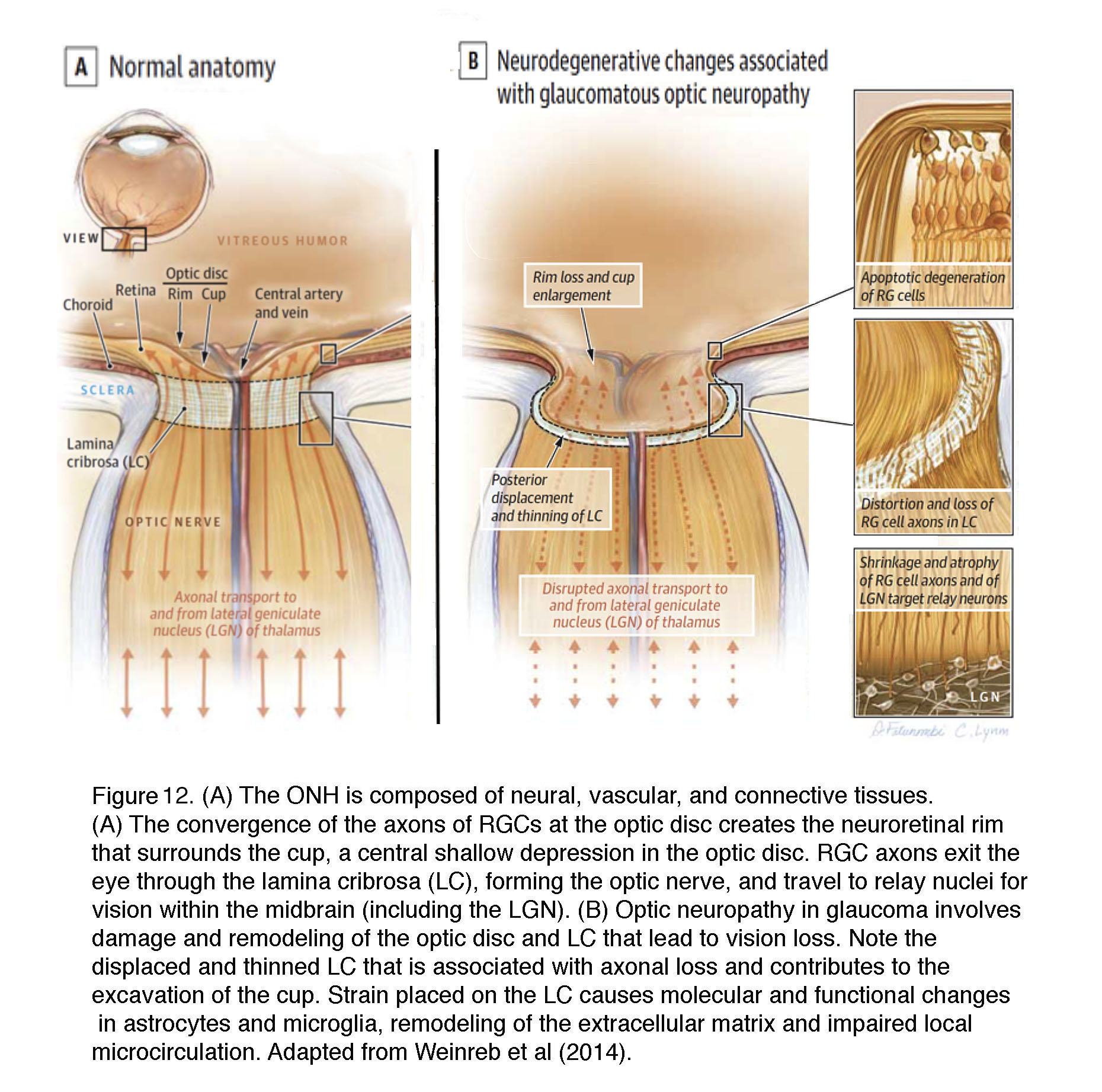 图12。(A)视神经头(视盘)由神经、血管和结缔组织组成。(A)视盘处视网膜神经节细胞(RG)轴突的汇聚形成了视网膜神经缘,它包围着视盘中央浅凹处的视杯。RG轴突通过眼板(LC)离开眼睛,形成视神经,并在中脑(包括LGN)内传播视觉的中继核。(B)青光眼视神经病变包括视盘和LC的损伤和重塑,从而导致视力丧失。注意与轴突损失相关的LC移位和变薄,并有助于杯的挖掘。置于LC上的应变导致星形胶质细胞和小胶质细胞的分子和功能改变,细胞外基质重塑和局部微循环损伤。改编自Weinreb等人(2014)。
图12。(A)视神经头(视盘)由神经、血管和结缔组织组成。(A)视盘处视网膜神经节细胞(RG)轴突的汇聚形成了视网膜神经缘,它包围着视盘中央浅凹处的视杯。RG轴突通过眼板(LC)离开眼睛,形成视神经,并在中脑(包括LGN)内传播视觉的中继核。(B)青光眼视神经病变包括视盘和LC的损伤和重塑,从而导致视力丧失。注意与轴突损失相关的LC移位和变薄,并有助于杯的挖掘。置于LC上的应变导致星形胶质细胞和小胶质细胞的分子和功能改变,细胞外基质重塑和局部微循环损伤。改编自Weinreb等人(2014)。
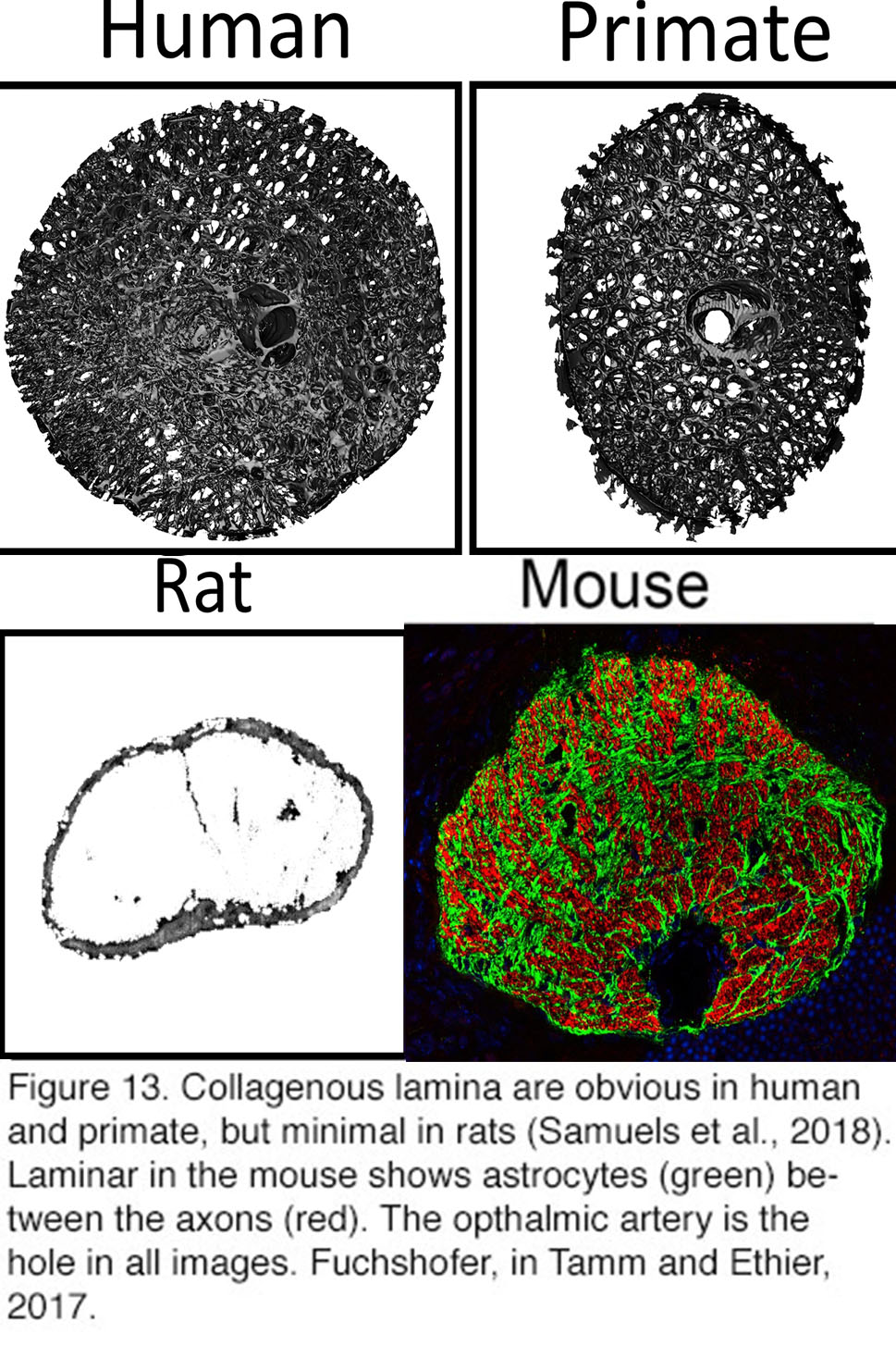 图13。胶原层在人类和灵长类动物中很明显,但在大鼠中很少。小鼠的椎板显示轴突(红色)之间的星形胶质细胞(绿色)。眼动脉是所有图像中的空洞。摘自Rudolf Fuchshofer,《Tamm and Ethier》,2017(141)。
图13。胶原层在人类和灵长类动物中很明显,但在大鼠中很少。小鼠的椎板显示轴突(红色)之间的星形胶质细胞(绿色)。眼动脉是所有图像中的空洞。摘自Rudolf Fuchshofer,《Tamm and Ethier》,2017(141)。
灵长类动物的筛板是由大约10个相互连接的ECM束组成的特殊区域,由I型胶原、III型胶原、IV型胶原和弹性蛋白组成。这些血管束由星形胶质细胞、小胶质细胞、成纤维细胞和广泛分支的血管供应组成,所有这些都可能相互作用。小的ECM细胞粘附可能持续数十秒,而局灶性粘附、细胞内肌动蛋白细胞骨架和ECM蛋白(纤维连接蛋白、肌腱蛋白、胶原蛋白等)之间的联系可在几分钟到几小时内形成(78)。在正常眼压(~15毫米汞柱)下,ONH所经历的机械应变在5% - 6%之间,而眼压升高到50毫米汞柱时,机械应变可增加到8 - 10%(79)。通常情况下,5-10%的细胞拉伸足以显著影响组织和器官的钙水平、细胞骨骼组织和组织生理学。生物力学建模和三维重建研究指出,ONH结构的粘弹性是ONH对眼压变化的动态响应的决定因素。特别是十字形ECM板被视为机械应变/压缩最弱的接触点(80)。压力的增加使ONH变硬,但尚不清楚更硬的板层是否更能抵抗机械应力。一个有用的类比可能是肥厚(纤维化,僵硬)的心脏,它允许更强的泵送对抗高背压,但最终对心血管健康有害。
必须指出的是,由于ONH的各向异性,以及不同几何形状的子区域在响应相同的IOP刺激时经历不同的张力的可能性,我们对影响RGC轴突的应变类型的理解仍然不完整(78)。眼压和视神经之间的压差大小尚不清楚,它是否受脑脊液压力的影响,巩膜组织的拉伸是否以及如何导致RGC损伤,某些RGC类别(哪些?)是否对IOP有选择性敏感/抵抗,为什么视网膜胶质细胞对IOP升高如此敏感,在中度眼压升高时,缺血压缩的作用是什么(如果有的话),哪些信号通路介导了高眼压的急性和慢性效应。这些通路可能通过op依赖性的促生存生长因子(BDNF, NT-3/4, NGF, CNTF)的顺行和逆行转运障碍、免疫基因表达的改变、局部趋化性、细胞- ecm相互作用和线粒体连接(81,82)。压力有可能通过机械传感器机制(如机械敏感TRPV4和TREK-1通道和整合素;(见下文),它介导了对巩膜拉伸、ONH压缩、张力和剪切的反应。在不同细胞类型(RGCs,胶质细胞,脉管系统)中这些机械转导器的激活可能决定了轴突和非轴突损伤的相对平衡,以及青光眼个体(和物种)之间神经炎症的重要性。
许多涉及轴突累及青光眼的研究都是在视神经挤压模型中进行的,而这些模型与压力诱导的轴突损伤没有直接关系。对原发性开角型青光眼(微珠诱导)和继发性闭角型青光眼(DBA/2J)动物模型的研究清楚地表明,压力升高会损害轴突运输(61),但它对神经退行性变的重要性仍有待证实,因为运输的长期减少不会影响乳头水肿等轴突疾病的RGC存活。此外,许多类型的RGC可以容忍运输的生存促进因子的损失,而缺乏持续的RGC存活的视网膜补充神经营养因子反驳了其损失的必然后果轴突运输受阻的动物。此外,DBA/2J小鼠(9-12个月)轴突运输的改变往往比树突重塑(1-3周;(55))、胶质反应性(3 ~6个月)和视神经信号缺陷(~6个月)(72,84,85)。观察到的顺行转运在逆行转运之前就已经被破坏(83),与大多数生物力学模型中预测的ONH收缩的关键作用相矛盾。相反,最近的研究表明非轴突机制的关键作用包括RGC活性的生理缺陷(55,83)和炎症信号(35,81)。
另一个反对青光眼中筛板的必要作用的重要论点是由进化提供的。层流ECM板在猪、猫、豚鼠、树鼩和狗中发育良好,但在啮齿动物中不存在。在大鼠和小鼠中,通过RGC轴突(约50,000小鼠)的结构和代谢支持由“胶质层”提供,由星形胶质细胞组成,在轴突束周围形成管(38,86)(图13)。因为这些动物仍会因眼压升高而发展成青光眼,所以无论是ECM板还是筛板都可能不是压力诱导的轴突损失所必需的。
眼压会导致大脑神经病变
青光眼损害不仅限于眼睛,还包括从视网膜到大脑的整个视觉通路。眼压增加导致空间视力丧失(87),同时降低外侧膝状核(LGN)的P层、k层和m层的大小、数量和代谢活性水平,而且与细小细胞细胞相比,巨细胞层的细胞明显更容易受到损伤。此外,初级视觉皮层的4C层也会发生变化(88)。因此,青光眼的再生策略将必须确保轴突的生存和再生,可能通过刺激视网膜受体传入(89)。这将需要注意青光眼相关的中脑RGC轴突终末变化和神经节细胞投影的视网膜位特异性(61)。
青光眼对巩膜和角膜的影响。
巩膜是一种密集的(400-1000 m厚)结缔组织,主要由胶原蛋白和弹性蛋白纤维组成,并含有产生ecm的成纤维细胞。它从角膜延伸到视神经,提供视网膜的机械支撑和眼外肌的插入。它与“乳头周围”区域内的板状膜接触(图12A),该区域因IOP而扩张,从而抵抗IOP。因此,巩膜可被认为是生物力学活跃的系统,由于IOP和由调节/收敛引起的肌肉收缩而产生张力,而这些张力又可从巩膜传递到相邻的镜板、视神经和提供ONH的血管系统(78,80)。我们知道机械压力调节眼睛的组成、生长和大小(90)部分通过巩膜MMP-2/TIMP2酶的活性(91)。与ONH和TM组织类似,随着年龄的增长和青光眼患者,巩膜变硬变薄,这可能会损害其承受iopo诱导的压力和保护更顺应性的视网膜和轴突的能力。
眼压依赖性视力下降的风险增加与角膜中央厚度变薄和粘弹性阻尼(“角膜迟滞”)有关(40)。这可能表明青光眼涉及眼内所有组织的分子机械转导和生物力学特性的改变。青光眼的主观视力丧失通常是由眼压迅速升高引起的角膜水肿引起的。不幸的是,人们对角膜上皮/内皮细胞如何感知压力和拉伸知之甚少(40)。
青光眼中的神经炎症:神经胶质细胞的重要功能。
压力依赖性视网膜胶质细胞的激活是青光眼的一个核心特征,但有时被忽视。在健康的视网膜,Muller细胞(图10,M,蓝色细胞)提供必要的代谢、渗透支持,并通过清除细胞外谷氨酸维持兴奋-抑制平衡。星形胶质细胞(图10,As,绿色)维持ONH结构,支撑轴突,维持血视网膜屏障。小神经胶质细胞(图10,Mi,黑色)调节适当的突触形成,吞噬和免疫信号传导,而少突胶质细胞(图10未显示)视神经中从椎板到中脑的RGC轴突髓鞘化。
青光眼胶质细胞对物理创伤、病理性肿胀和神经退行性疾病引起的神经损伤的反应模式与大脑对应细胞的反应模式相一致。眼压升高引起视网膜神经胶质基因表达、神经胶质递质释放的深刻变化,并损害其在一氧化氮、细胞因子、趋化因子、免疫样分子(如补体、CX3CL1、TNFα、IL-1β)和活性氧(ROS)信号中的作用(35,85)。短暂而轻微的IOP升高可能在RGC/轴突明显损伤之前诱导Müller细胞、星形胶质细胞和小胶质细胞的反应性激活。一个突出的例子是细胞骨架、ECM蛋白和结构蛋白(胶质酸性纤维蛋白GFAP、波形蛋白、胶原、层粘连蛋白和肌腱蛋白)的胶质依赖性变化和蛋白多糖的下调(92)。高眼压也是视网膜胶质细胞代谢组重塑的基础,包括胶质细胞谷氨酸摄取和谷氨酰胺生物合成的下调,这可能会影响神经元的兴奋性和功能。压力诱导的促炎神经胶质信号的阳是由神经胶质释放的保护性神经营养CNTF和/或BDNF因子和JAK/STAT3信号的营养保护的阴。
星形胶质细胞在视神经头和青光眼
与大脑相比,视网膜中的星形胶质细胞较少,但仍占灵长类ONH细胞的约50%。它们的视网膜功能包括维持血管张力、ECM产生和结构支持(93)。星形胶质细胞通过ATP、前列腺素、血管紧张素、腺苷和EET(环氧二碳三烯酸)的释放调节神经活性和神经血管耦合。此外,星形胶质细胞作为ECM的主要产生者,有助于青光眼中筛板的机械顺应性及其重塑。哺乳动物的视网膜含有具有独特分子和解剖学特征的星形胶质细胞。I型gfap阳性星形胶质细胞位于人ONH的前层和无髓鞘区域(图10和14中绿色,图15A中红色),覆盖ECM束和嵴孔,包住轴突束,包围血管,并介导轴突和毛细血管之间的代谢物转移(图10和14中绿色)。另一种亚型,GFAP-阴性和α-SMA(平滑肌肌动蛋白)-阴性的2型星形胶质细胞主要存在于ONH的有髓鞘后层区(94),在那里它们与最近发现的GFAP- α-SMA+“筛层细胞”的一个亚群共存,该亚群可能通过胶原、弹性蛋白和纤维连接蛋白的功能失调合成削弱ONH(95)。在啮齿类动物中,星形胶质细胞是根据形态学而不是分子标准分为原生质和纤维亚型(92)。与哺乳动物一样,啮齿动物的星形胶质细胞分布在视网膜上,以调节神经血管功能(图10,图15b面板E),并发出包裹和分隔轴索束的细突起。与ONH相反,啮齿动物星形胶质细胞也在“胶质层”内提供结构支持(图13)。 However, in animals with avascular retinas (such as rabbits), astrocytes are absent from areas that lack blood vessels.
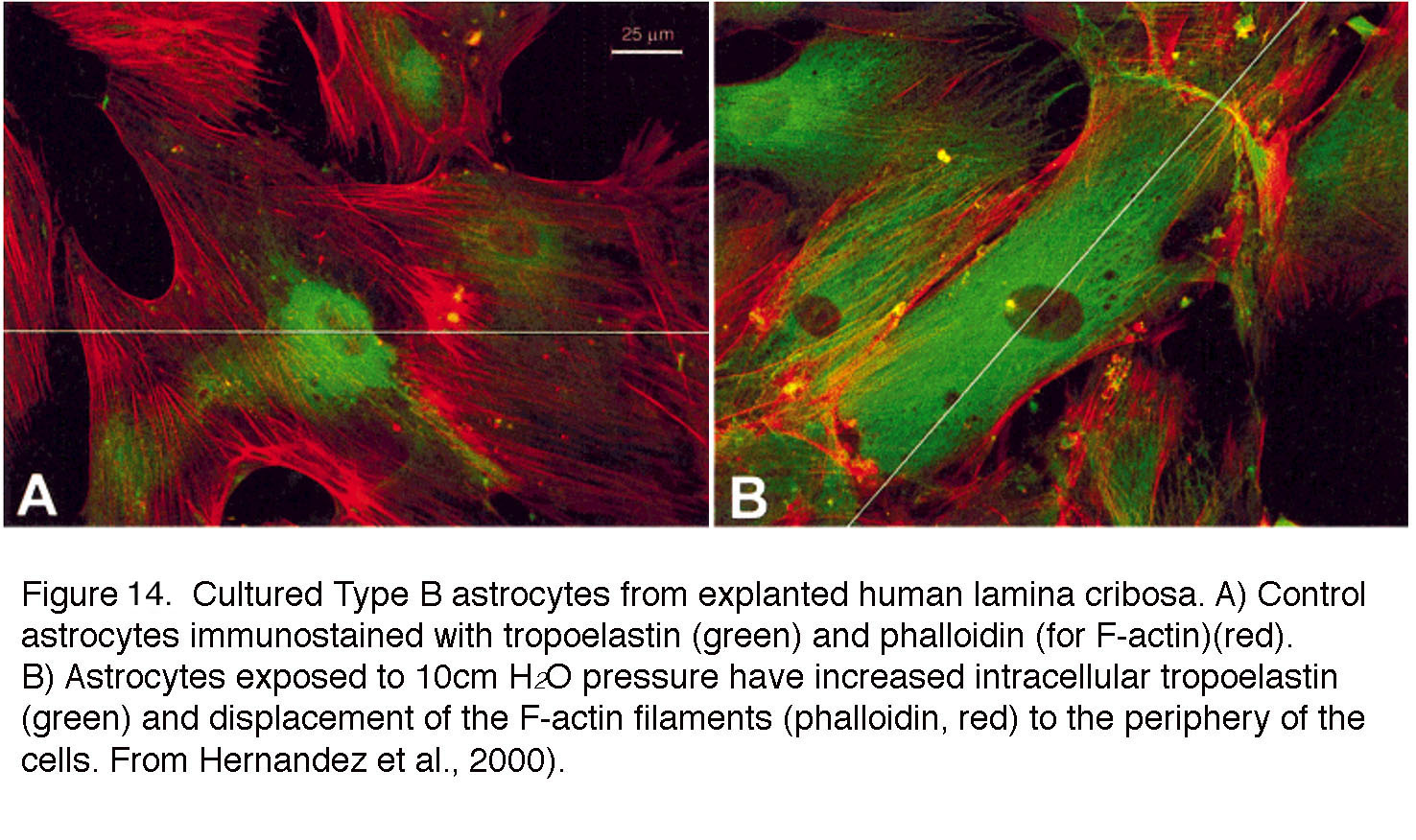 图14。体外培养的人筛板B型星形胶质细胞。(A)用对偶弹性蛋白(绿色)和phalloidin (F-actin,红色)免疫染色的对照星形胶质细胞。(B)暴露于10cm H2O压力下的星形胶质细胞细胞内对流层弹性蛋白(绿色)增加,f -肌动蛋白丝(phalloidin,红色)移至细胞外围。摘自Hernandez等,2000(98)。
图14。体外培养的人筛板B型星形胶质细胞。(A)用对偶弹性蛋白(绿色)和phalloidin (F-actin,红色)免疫染色的对照星形胶质细胞。(B)暴露于10cm H2O压力下的星形胶质细胞细胞内对流层弹性蛋白(绿色)增加,f -肌动蛋白丝(phalloidin,红色)移至细胞外围。摘自Hernandez等,2000(98)。
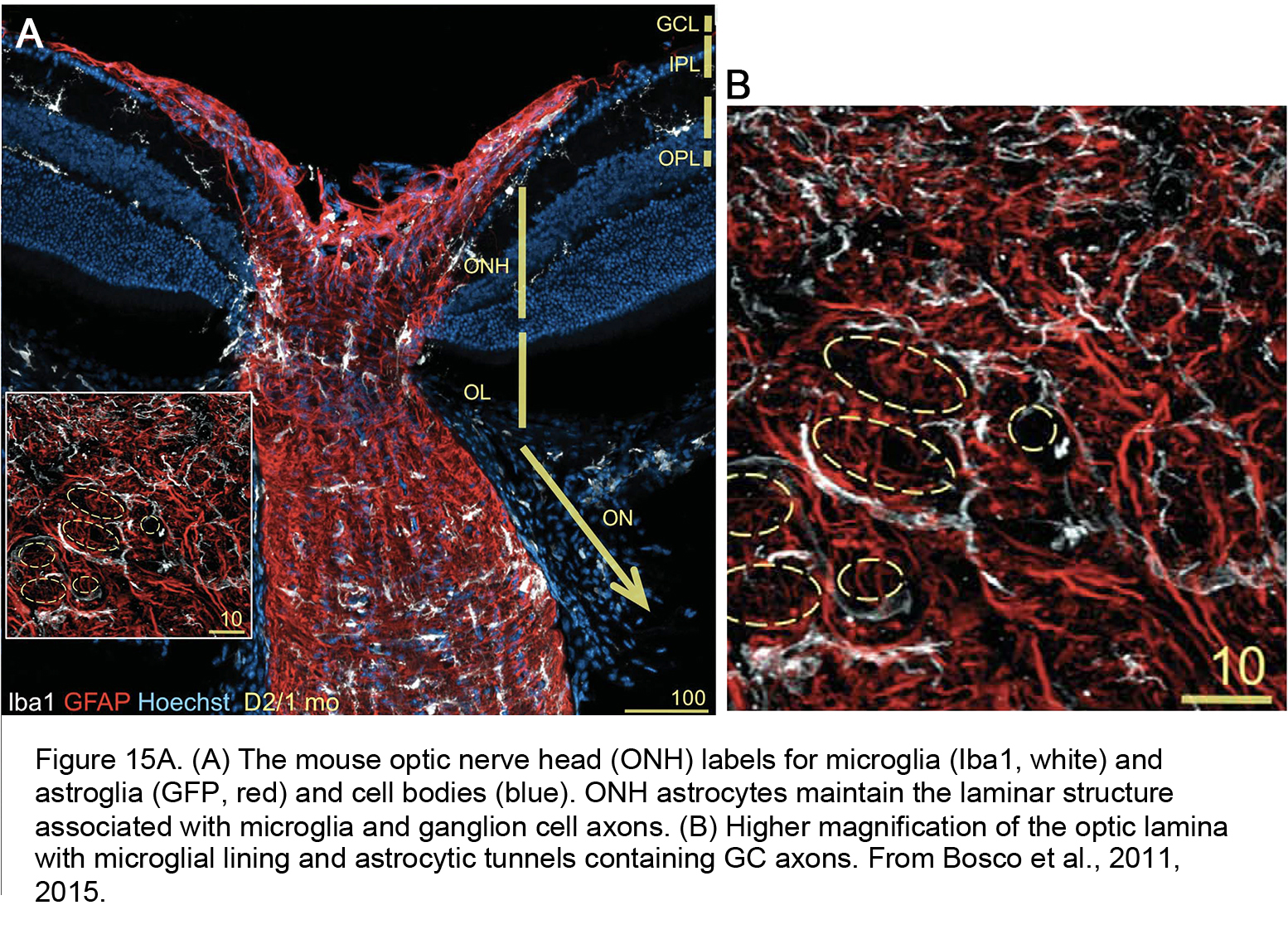 图15。(A)小鼠视神经头(ONH)小胶质细胞(Iba1,白色)和星形胶质细胞(GFP,红色)和细胞体(蓝色)的标签。ONH星形胶质细胞维持与小胶质细胞和神经节细胞轴突相关的层状结构。(B)具有小胶质衬里和星形细胞隧道包含气相色谱轴突的光学层。来自Bosco et al., 2011(84)。
图15。(A)小鼠视神经头(ONH)小胶质细胞(Iba1,白色)和星形胶质细胞(GFP,红色)和细胞体(蓝色)的标签。ONH星形胶质细胞维持与小胶质细胞和神经节细胞轴突相关的层状结构。(B)具有小胶质衬里和星形细胞隧道包含气相色谱轴突的光学层。来自Bosco et al., 2011(84)。
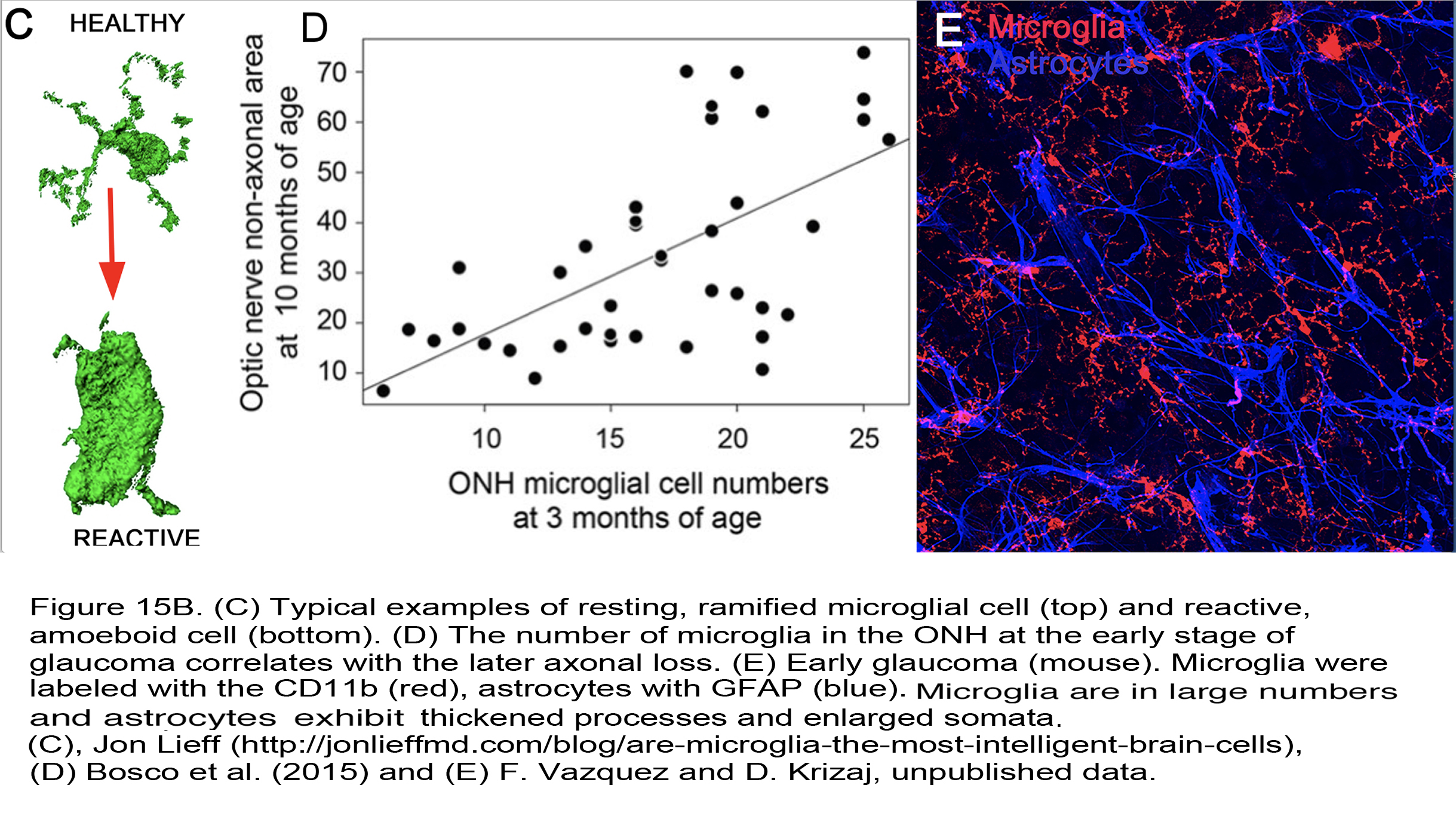 图15 b。(C)静止的分枝小胶质细胞(上)和反应的变形虫细胞(下)的典型例子。(D)青光眼早期ONH中的小胶质细胞数量与青光眼后期轴突损失相关。(E)早期青光眼(小鼠)。小胶质细胞用CD11b标记(红色),星形胶质细胞用GFAP标记(蓝色)。细胞有肥大的迹象(突起增厚和体块增大)。图片来自Jonn Lieff (142) (C), Bosco等人(2015)(109)(D), F. Vazquez和D. Krizaj,未发表数据(E)。
图15 b。(C)静止的分枝小胶质细胞(上)和反应的变形虫细胞(下)的典型例子。(D)青光眼早期ONH中的小胶质细胞数量与青光眼后期轴突损失相关。(E)早期青光眼(小鼠)。小胶质细胞用CD11b标记(红色),星形胶质细胞用GFAP标记(蓝色)。细胞有肥大的迹象(突起增厚和体块增大)。图片来自Jonn Lieff (142) (C), Bosco等人(2015)(109)(D), F. Vazquez和D. Krizaj,未发表数据(E)。
相邻的星形胶质细胞通过同源体(如Cx 43)和异型缝隙连接耦合,这些缝隙连接在胶质网络中重新分配钙离子和代谢物,但也通过释放神经活性化合物调节神经元功能(96,97)。这些细胞是纤维连接蛋白的主要来源,但在体内和体外条件下,也会上调许多ECM蛋白,以应对机械应激源(压力、拉伸)(例如,肌腱蛋白、层粘连蛋白、胶原蛋白、弹性蛋白和血栓反应蛋白)(例如,图14)以及与细胞运动和迁移相关的基因(凝胶蛋白、LIMBP1、板周蛋白)和下调整合素基因(99,98)。
机械压力会极大地改变视网膜星形胶质细胞的基因表达、形态和功能,并将细胞推入一种反应状态astrogliosis表现为椎板的炎症和纤维化,并与ONH结构减弱、血供受损和轴索损失相关(86,98,99)。青光眼ONH中的星形胶质细胞增厚、钙依赖性肥大和增殖(86,99)。与iopinduced astrogliosis相关的基因/蛋白质列表包括数百个编码细胞周期蛋白、丝裂原激活蛋白激酶(MAPKs)、骨形态发生蛋白(bmp)、内皮素受体(ET)的基因一个和等B),基质金属蛋白酶(MMPs), JAK/STAT3转录,细胞骨架蛋白(GFAP,波形蛋白,胶原蛋白,层粘连蛋白和肌腱蛋白),细胞粘附成分,信号转导分子(NF- kb, STAT5, MCP-1, glypicans),免疫/补体系统(C1q,细胞因子如IL-6, LIF),骨膜素,吞噬相关基因Mac-2,和/或星形胶质炎性体的NF-κ b依赖性组装(81,100,101)。视网膜星形胶质细胞释放的细胞因子TGF-β2抑制mmp依赖的ECM降解,同时促进胶原、弹性蛋白和纤维连接蛋白的表达(92),这可能导致更硬的层板和ONH损伤。压力激活星形胶质细胞的另一个特征是迁移反应,使它们能够爬出筛层板(102)。
虽然我们不知道眼压升高是如何诱导星形胶质细胞分化的,但生理学研究表明机械应激和星形胶质细胞[Ca2+]i(97)之间有直接联系。物理力诱导视网膜星形细胞膜的跨膜电流(103),触发星形细胞和Müller胶质细胞之间的钙波传播,并刺激促炎介质的释放。因此,视网膜星形胶质机械转导最好是在与相邻小胶质细胞、Müller细胞、微血管系统和rgc的反馈循环的背景下进行观察(104)。
青光眼中的小胶质细胞和单核细胞
青光眼涉及适应性和先天免疫机制,视网膜小胶质细胞——构成先天免疫系统——是机械应激最早的哨兵之一。在健康视网膜中,小胶质细胞(Mi;图10中的黑色)参与局部环境的动态监测和局部突触接触的形成,但这种活动在与青光眼小胶质细胞相关的促炎表型中显著改变。这些细胞的特征是增殖、过程收缩和补体、toll样受体(TLR2-4)、TNFR1α和β受体、一氧化氮合酶、热休克蛋白(HSP27、HSP60、HSP72)、趋化因子(IL-8)和细胞因子(IL-1β、IL-6、CCL2、TNFα)信号的上调,以及抗炎TGFβ信号的抑制(35,105)。在疾病早期,金属蛋白酶(MMP-1-3)的释放可能改变ECM并使其不稳定。Vetter实验室最近的一项重要研究表明,小胶质细胞在发育过程中选择存活的rgc方面发挥着关键作用(106)。在ONH中,首先在早期轴突损伤区域检测到微胶质细胞激活(图15A,图15B),这可能是对rgc的fractalkine (CX3CL1)输入丢失的响应(107)和与Müller胶质细胞的串音改变(108)。在DBA/2J小鼠青光眼模型中,二甲胺四环素(一种小分子微胶质激活抑制剂)和波生坦(一种小胶质内皮素(ET1/2)受体抑制剂)都能增加神经节细胞存活(81,109)。
青光眼视网膜免疫激活的另一个重要特征是血视网膜屏障的通透性丧失,这允许来自体循环的活化单核细胞和树突状细胞的入侵(81,105)。青光眼与经典补体通路和可选补体通路中的小胶质细胞变化有关,“可选”C1q复合体(38)可能促进了“经典”C3转化酶的激活,该酶激活突触修剪和体细胞吞噬。另一个重要的角色是细胞因子IL-1β和TNFα,它们促进青光眼视网膜的RGC凋亡(105)。一只眼的眼压升高会触发两只眼的小胶质细胞激活,这证明了青光眼的免疫机制与弥漫性全身因子的扩散有关。与此一致的是,在高血压眼的血管周围形成活化的小胶质细胞簇(110)和啮齿动物青光眼的树突状和突触结构不仅可以通过抑制C1q蛋白或删除其基因,还可以通过阻止巨噬细胞/单核细胞的压力诱导迁移(60,81)来保护。此外,小胶质细胞可能对机械释放的趋化因子产生反应,如Müller胶质细胞中的单核细胞趋化蛋白(MCP-1)(111)和神经节细胞中的fractalkine (CX3CL1)(107)。
Muller细胞
Muller细胞(米;图10中的蓝色)是放射状胶质细胞,从视网膜外侧延伸到最内侧。它们包围着每一个神经元,几乎调节视网膜功能的每一个方面,从能量代谢到体积调节,血液流动,神经递质的摄取和循环,以及免疫信号(112)。它们的膜性质主要由钾离子通道Kir 4.1依赖的内整流所支配,它淹没了其他离子通道的贡献。Müller尾足有助于维持血视网膜屏障的神经胶质血管单元(图16,对照),也分泌基底膜,形成视网膜与IOP相互作用的内限制膜。端足强烈表达拉伸激活的TRPV4通道,因此对拉伸、戳和体积变化非常敏感(27,113)。
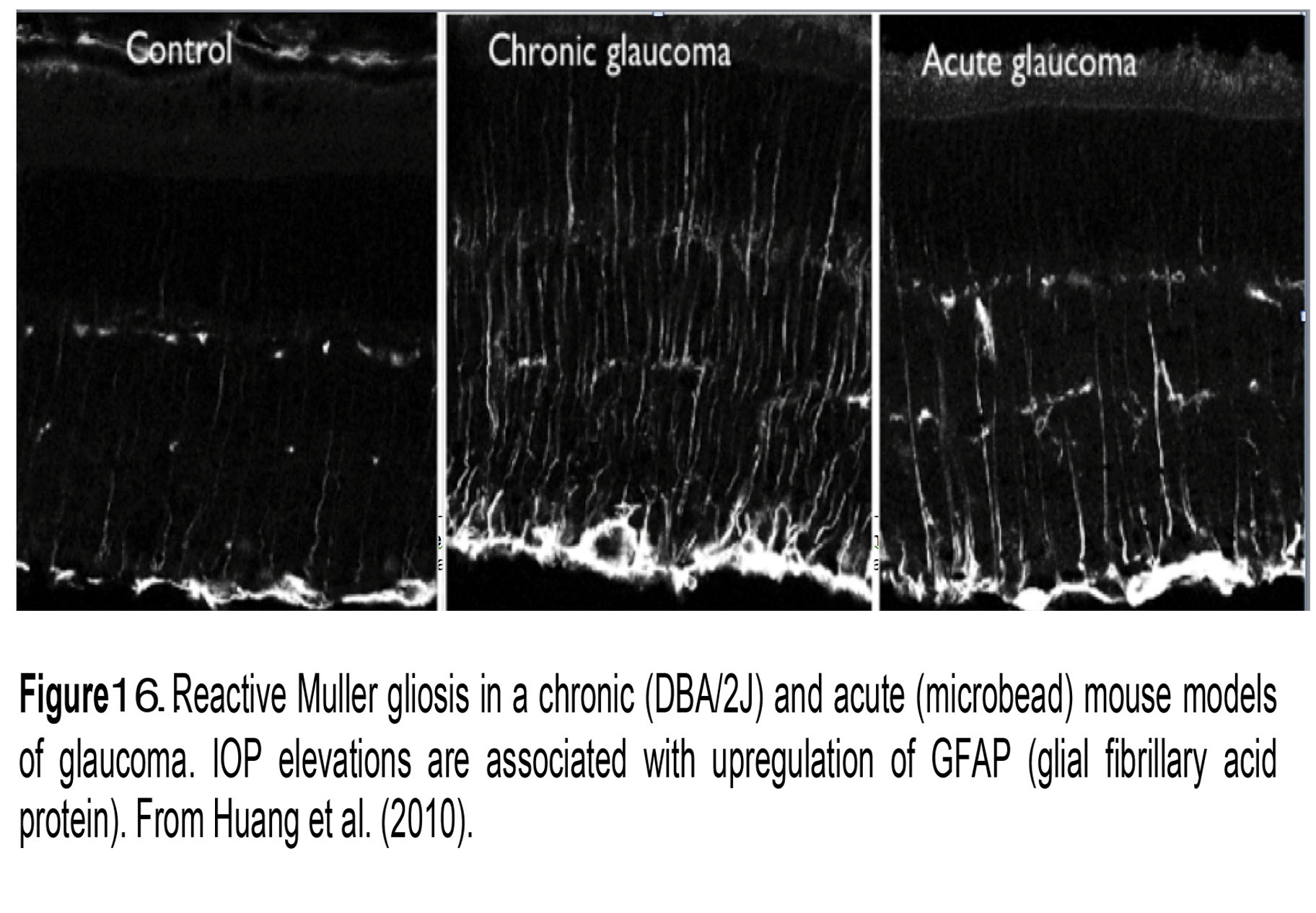 图16。慢性(DBA/2J)和急性(微珠)小鼠青光眼模型的反应性Müller光泽。眼压升高与GFAP(胶质纤维酸蛋白)的上调有关。来自黄等人(2010)。
图16。慢性(DBA/2J)和急性(微珠)小鼠青光眼模型的反应性Müller光泽。眼压升高与GFAP(胶质纤维酸蛋白)的上调有关。来自黄等人(2010)。
Müller神经胶质细胞是首批对IOP升高有反应的视网膜细胞之一(85)。急性和慢性IOP升高将其推入反应性状态(反应性胶质细胞增生),这通常通过免疫标记视网膜的中间丝GFAP可见(图16;慢性和急性青光眼)。缺乏GFAP的小鼠可能对机械损伤更敏感(114),可能是因为这些细胞骨架元素抵抗机械应力。在反应模式下,Müller细胞可能通过释放一氧化氮、TNFα/IL-1β/NF-κB/mTOR信号和释放细胞因子MCP-1来调节血管通透性和神经元存活(35,111)。受压力影响的Müller信号传导的其他方面包括谷氨酰胺合成/谷氨酸摄取的下调,细胞骨架蛋白的上调,以及水通道AQP4(水)通道、钙通道(赖氨酸受体)、补体蛋白(C1q)和晚期糖基化终产物(RAGE)的表达变化(72,115,116)。
内皮细胞,周细胞和局部血液供应
ONH的血液供应来自眼动脉,眼动脉分支进入视网膜中央动脉,灌注神经纤维层(图10)和两个睫状体后动脉。静脉血流入经视神经的中央静脉(图12)。较大的眼压升高可诱导缺血,从而影响葡萄糖、氧和乳酸通过毛细血管压迫的扩散。然而,即使是很小的眼压升高(3-5 mm Hg)也可能导致缺氧、活性氧和缺氧诱导因子(如HIF-1α和HIF-2α)的积累、星形胶质谷胱甘肽含量的下调和自噬体的诱导(117)。
未来的研究挑战
钙的重要性
在过去的几十年里,人们一直在努力确定青光眼视网膜损伤的最终共同机制。就rgc而言,这些机制包括内质网应激、促凋亡级联的激活以及轴突和非轴突间室内钙稳态的失调(35,39)。钙作为第二信使,在全身的细胞生理和病理中起着至关重要的作用,几乎每一种与青光眼相关的细胞类型,以及青光眼的每一个阶段,似乎都与Ca2+调节失调有关,这并不奇怪。青光眼中与病理性Ca2+稳态相关的来源和途径可能包括质膜内的离子通道和细胞内腔室如内质网(ER)、线粒体和溶酶体释放的钙。不难想象,[Ca2+]i的这种变化促进TM收缩力,驱动视网膜胶质细胞的炎症激活,促进rgc对继发性损伤的易感性。
体外实验表明,机械应力触发视网膜神经节细胞、微血管内皮细胞、睫状体非色素细胞、胶质细胞和小梁网(51,11,118 -120)中的[Ca2+]i升高,而外部介质中的Ca2+去除或钙螯合阻断拉伸和肿胀诱导的Ca2+内流和拉伸诱导的细胞骨架重构。有证据表明青光眼RGCs中[Ca2+]i的增加发生在树突和体细胞中(121),这可能解释了青光眼RGCs中内质网应激和Ca2+依赖性信号(即早期基因(c-fos, JNK2/3, CREB),蛋白酶,caspases和MAP激酶)的增加。另一种Ca2+依赖的机制涉及钙酸钙的构成激活,钙+依赖的酶促进半胱天pasase激活和蛋白质降解。钙调磷酸酶的抑制在啮齿动物神经节细胞损伤模型中具有保护作用,可能是通过Ca2+激活的磷酸酶钙调磷酸酶(71)。然而,钙调神经酸-钙pain通路也有可能促进轴突再生(122)。
上述发现表明,靶向压力诱导钙流入可能有助于缓解视网膜病理。这种保护作用在非生理条件(视神经横断/挤压)下可见,电压和谷氨酸依赖性钙流入的阻断对RGC存活和轴突变性具有很大的保护作用。我们假设,阻断对压力敏感的钙透性通道或其生物力学结果(如张力拉伸)可能会导致更显著的高血压眼损伤改善。
转导的重要性
一个关键的要求是确定眼压感知、房水动力学和神经退行性变之间的分子联系,否则动物和人类的青光眼就无法被理解。为了达到这一目的,我们需要更好地了解眼内机械冲击的实际位置,并识别由前眼和后眼细胞感知的机械压力源(拉伸应变、剪切流、压缩等)。对青光眼动物模型和孤立细胞的研究表明,大多数与青光眼相关的细胞类型(小梁网细胞、视网膜胶质细胞和RGCs)对压力的反应是快速启动的电信号和钙信号(~毫秒到~秒),而持续暴露于机械应激源往往会引发青光眼损伤的特征表型。除了对压力传感器分子进行表征外,确定眼细胞如何忽略稳态眼压,以及它们如何响应和补偿由眨眼、咳嗽、眼部摩擦、昼夜周期、肿胀、眼外伤和运动(如游泳镜、瑜伽练习)引发的短暂眼压波动也很重要。为了识别分子机制,我们现在可以建立从细胞机械感应研究中获得的大量信息。
机械生物学是生物学和生物工程相结合的一个新兴领域,研究物理力和细胞力学特性的变化如何促进细胞的发育、分化、增殖和疾病。它的共同主题是,每个细胞通过使用专门的蛋白质将机械输入转化为电子或生化信号,这些蛋白质控制着从纳米尺度到宏观尺度几乎每一个组织层面的机械稳定性和机械信号传输(123,124)。这些机械转导机器在细胞-细胞和组织-组织网络水平上施加调控约束,但有些细胞比其他细胞更擅长机械转导,因此对机械力更敏感,并在疾病中调节失调。经常被引用的机械转导出错的例子是由机械敏感蛋白突变引起的听力和平衡疾病,但还有许多其他的例子是由压力和拉伸增加引起的骨、软骨、肺、膀胱、免疫系统、心脏和眼睛疾病(125)。例如,青光眼和近视可以被认为是机械转导疾病,反映了眼睛细胞对特定类型的局部机械应激的功能障碍反应。
许多蛋白质家族通过构象变化来响应机械力。机械敏感蛋白包括局部粘附蛋白、细胞-细胞粘附、整合素和ECM蛋白、受体酪氨酸激酶、g蛋白偶联受体、细胞骨架蛋白,重要的是,在缺乏化学配体的情况下,细胞膜曲率和拉伸的微小变化会激活离子通道(124-126)。这些机械敏感通道的进化是为了感知质膜的亚微米位移,但在初级结构、离子选择性、电导和药理学方面表现出巨大的差异(图17)。它们被分为Na+-渗透性(ENaC/degenerins)、K+-渗透性(TREK, TRAAK)和非选择性阳离子(TRP和piezo)通道家族,它们表现出异构体特异性的机械阈值、时间过程、对持续刺激的适应,以及它们与细胞骨架的连接方式和对翻译后调制的响应方式的差异。
从概念上讲,机械通道的功能是带通滤波器,可以传输特定的振幅和类型的机械应力(例如,压缩、张力、膨胀、剪切流等),范围从温和的触摸、压力、纹理、振动、声音、剪切流到物理创伤和疼痛。最近的研究表明,眼中的青光眼相关组织(TM、睫状体、神经元、胶质细胞、角膜、内皮细胞)表达多种机械敏色氨酸TRP、piezo和串联孔隙K+通道(27,127 -129)。这些通道的药物抑制和遗传消融可改善与高眼压相关的炎症和退行性视网膜表型(119),提示可能在青光眼中起作用。
图17展示了香草色氨酸色氨酸(TRPV)和TREK (TWIK-1相关K+)机械传感器的基本拓扑结构。TRPV4是香草色氨酸色氨酸家族的一员,它介导Na+和Ca2+离子的流入,以应对各种各样的机械和化学刺激(130,131),包括压力、拉伸、肿胀、戳和体温(34 - 37℃)。它在约60-70%的小鼠rgc中表达,在100%的Müller细胞中表达(图18A),在大部分的小梁和角膜上皮细胞中表达(70,113,132),一些rgc与同源TRPV1通道共享TRPV4表达(图18B)(70,128)。有趣的是,该通道似乎在对青光眼不太敏感的神经元类别(光感受器,双极,无分泌细胞;图10)和缺乏TRPV4的小鼠视网膜外功能无缺陷(图18C)。TRPV4通道通常通过n端锚结蛋白重复序列整合到细胞骨架网络中(图17A),并通过α2整合素分子和局部粘附与ECM网络连接。它们对压力敏感(129),驱动拉伸诱导的细胞骨骼重塑,并可能导致慢性眼压升高期间张力内稳态的丧失(51)。
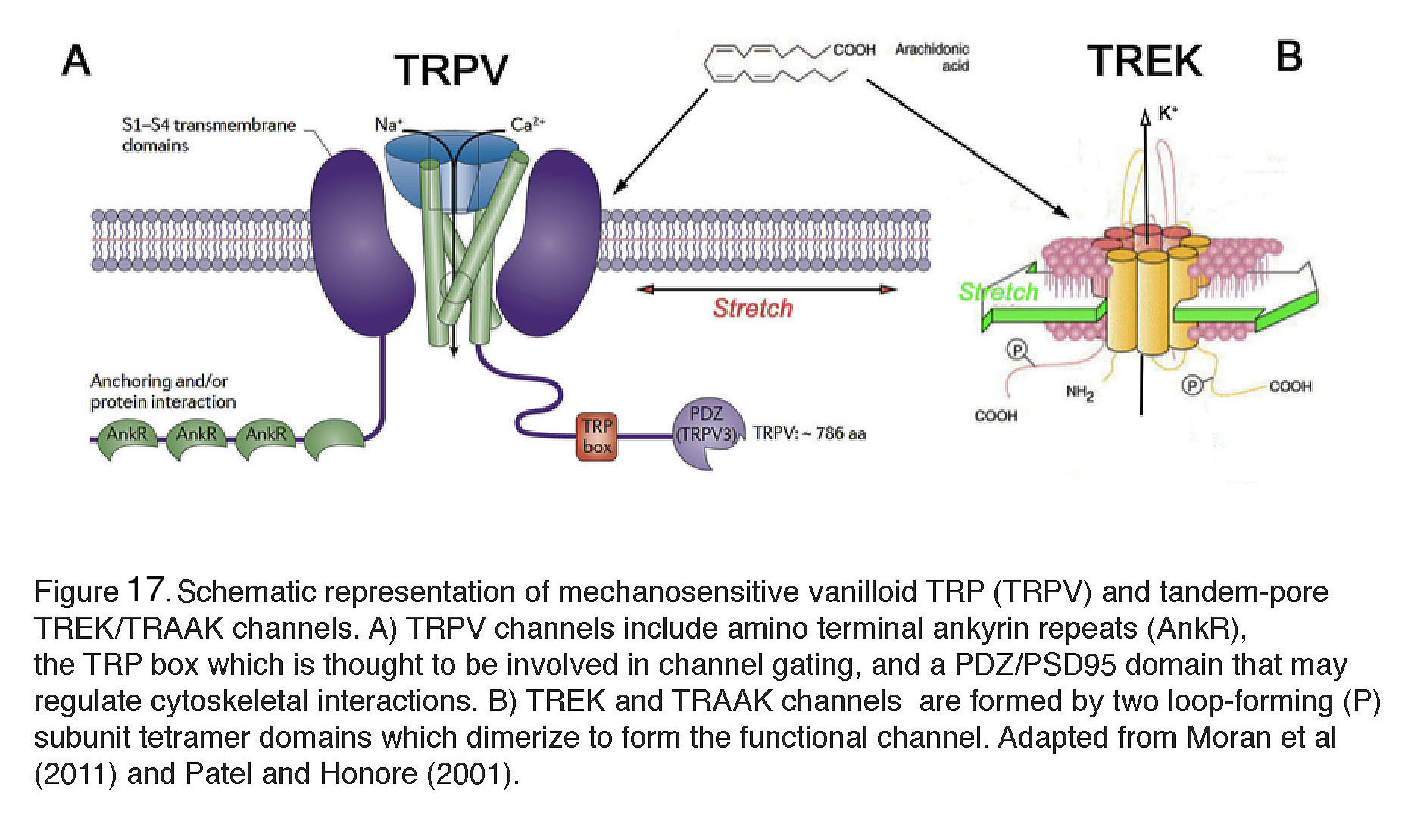 图17。机械敏香草质TRP (TRPV)和串联孔TREK/TRAAK通道示意图。(A) TRPV通道包括氨基末端锚定蛋白重复序列(AnkR),被认为参与通道门控的TRP盒,以及可能调节细胞骨架相互作用的PDZ/PSD95结构域。(B) TREK和TRAAK通道是由两个环形成(P)亚基四聚体结构域二聚形成功能通道。改编自Moran等,2011(143)和Patel和Honoré, 2001(144)。
图17。机械敏香草质TRP (TRPV)和串联孔TREK/TRAAK通道示意图。(A) TRPV通道包括氨基末端锚定蛋白重复序列(AnkR),被认为参与通道门控的TRP盒,以及可能调节细胞骨架相互作用的PDZ/PSD95结构域。(B) TREK和TRAAK通道是由两个环形成(P)亚基四聚体结构域二聚形成功能通道。改编自Moran等,2011(143)和Patel和Honoré, 2001(144)。
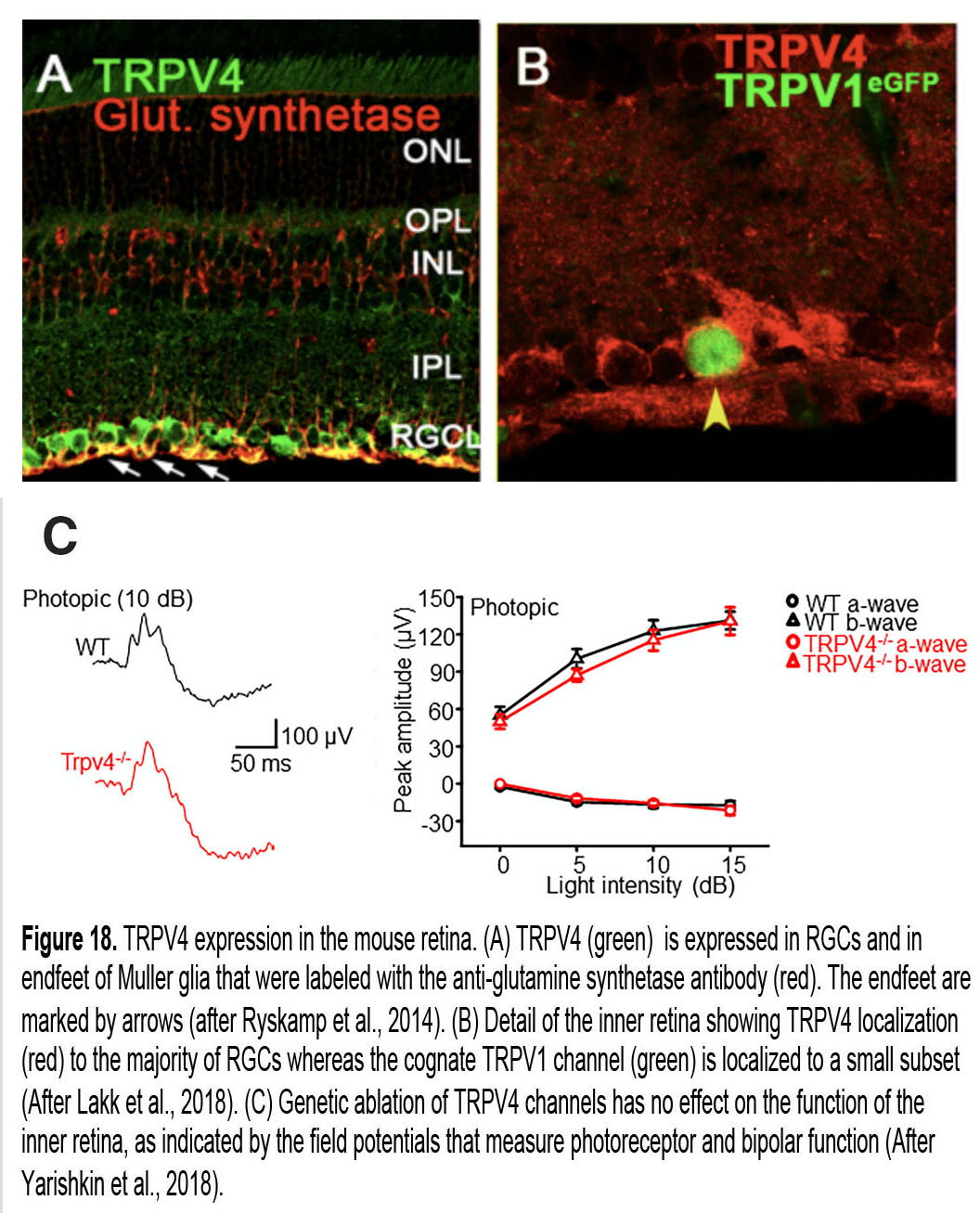 图18。TRPV4在小鼠视网膜中的表达。(A) TRPV4(绿色)在抗谷氨酰胺合成酶抗体(红色)标记的视网膜神经节细胞和Müller胶质细胞尾足中表达。端脚用箭头标出。Ryskamp等,2014(27)。(B)视网膜内部的细节显示TRPV4定位(红色)在大多数视网膜神经节细胞,而同源TRPV1通道(绿色)定位在一小部分。Lakk等人,2018(70)。(C) TRPV4通道的遗传消融对内视网膜的功能没有影响,测量光感受器和双极功能的场势表明。Yarishkin等,2018(145)。
图18。TRPV4在小鼠视网膜中的表达。(A) TRPV4(绿色)在抗谷氨酰胺合成酶抗体(红色)标记的视网膜神经节细胞和Müller胶质细胞尾足中表达。端脚用箭头标出。Ryskamp等,2014(27)。(B)视网膜内部的细节显示TRPV4定位(红色)在大多数视网膜神经节细胞,而同源TRPV1通道(绿色)定位在一小部分。Lakk等人,2018(70)。(C) TRPV4通道的遗传消融对内视网膜的功能没有影响,测量光感受器和双极功能的场势表明。Yarishkin等,2018(145)。
青光眼的许多眼部表现(眼压改变、钙负荷过重、反应性胶质细胞增生、细胞因子释放、微血管功能障碍、肌动蛋白上调、纤维连接蛋白分泌和RGC凋亡)可通过刺激RGCs、胶质细胞、内皮细胞、睫状体和小梁网细胞中的TRPV4通道进行药理学模拟(27,111,127,133)。TRPV4诱导的表型包括RGC变性(133)、反应性胶质细胞增生(27)、胶质细胞肿胀(113)和血视网膜屏障通透性增加(127,134),而缺乏TRPV4基因的小鼠似乎不受某些类型的机械和伤害性应激的影响(130),并没有出现青光眼(51)。胆固醇和多不饱和脂肪酸——青光眼的潜在危险因素——也调节TRPV4的激活(135)。
与色氨酸酶和压电通道激活相反,健康眼睛的机械内稳态是如何维持的?一种可能反对TRPV4过度激活的机械传感器候选者可能是K+渗透性的TREK-1通道。TREK-1对抑制TRPV4的刺激异常敏感(膜拉伸、肿胀、体温和花生四烯酸),经常与TRPV4共同表达(O. Yarishkin, F. Vazquez-Chona和d.k.,未发表的观察结果)。缺乏TRPV4通道的小鼠对机械应力不太敏感(131),而TREK/TRAAK通道切除的小鼠对机械力和局部pH值变化异常敏感(136)。根据该模型,TRPV4/piezo与TREK-1(及其同源TRAAK)通道的同时激活可能维持至少部分眼组织的张力内稳态。
摘要和结论
青光眼是全球不可逆失明的主要原因。该疾病有多种形式,但都涉及与视神经病变相关的前眼信号功能障碍。这种病无法治愈,目前也没有办法恢复失去的视力。这是一个问题,因为青光眼在晚期通常是无症状的,许多青光眼患者没有意识到自己患病,直到为时已晚才能进行有效治疗。因此,有危险因素的患者(即有家族史和/或非洲血统的老年人)定期进行筛查是很重要的。诊断后,治疗将是终生的,但需要严格遵守治疗方案(降低眼压滴眼液)。
青光眼是什么?最明确的答案是,我们仍然不知道,因为病理学可以反映多因素病理学和/或最终具有类似共同病理学的一系列疾病。有趣的是,老年人和青光眼患者的机械转导似乎在眼睛的所有组织(角膜、巩膜、TM、视网膜)中都受到了损害,这表明细胞产生和处理机械力的能力出现了广泛的功能障碍。大多数形式的青光眼似乎是遗传的,但很可能为了病理表现,多种蛋白质和/或脂质的突变/改变是必需的,而病因基因和突变的识别将极大地促进诊断和治疗。目前,只有5-10%的病例与特定基因有关。
降低眼压——目前主要的治疗方法——从滴眼液开始,对于难治性或晚期的病例,只能采用激光手术和切口手术。此时,抗青光眼药物主要针对介导少量液体流出的次要通路,有很大的改进空间,特别是针对小梁流出的治疗。另一个重点是保护视网膜神经元和胶质细胞不受压力的影响,这就需要阐明感知和传递机械压力的分子/细胞机制。具体来说,改善治疗需要研究科学家、临床医生和生物医学工程师:(i)确定风险因素、遗传易感性和构成眼睛前后细胞相互作用基础的分子机制之间的定量关系,(ii)开发标准化测试,以确定特定患者的视神经病变在“正常”衰老和青光眼之间的光谱中的哪个位置,(iii)开发用于早期诊断的新技术(如光学相干断层扫描);(iv)提高靶向TM的药物的疗效,同时消除副作用;(v)保护小梁网、视网膜神经节细胞和胶质细胞内的压敏靶点。这需要对神经保护治疗进行投资,但由于科学家们对RGC机制转导机制的分歧、临床试验的昂贵费用以及公认的神经保护药物最近的昂贵失败,这一投资一直受到阻碍。
未来治疗的主要目标必须是降低眼压,同时防止压力依赖性炎症和神经退行性变,这一方法可能包括直接靶向压力传感蛋白本身。不幸的是,尽管经过了几十年的密集研究,目前似乎对IOP的早期视网膜靶点(树突?somata吗?轴突?神经胶质吗?血管?“?)例如,越来越明显的是,神经胶质细胞是对高压最早的反应者之一,神经退行性变可以在IOP稳定后持续很长时间。因此,任何神经保护的尝试都必须考虑到独立于op的血管、神经胶质和巩膜机制。青光眼的预防很可能会随着对分子、昼夜节律、热量限制和运动参数的更好理解而得到改善,这些参数越来越被认为与眼部健康相关。 Another innovative front will definitely be on the technology side, as patients are equipped with real-time telemetric devices, noninvasive ONH measurements become routine, and diagnoses are provided by deep learning (artificial intelligence) algorithms (46). We live in exciting times that, having unveiled the key likely components and patterns of visual damage in glaucoma and old age, can only lead to glorious future where mechanotransduction mechanisms in individuals at risk for disease onset are understood, targeted and used for early diagnosis.
确认
由美国国立卫生研究院、Willard L. Eccles基金会、青光眼研究基金会、犹他大学技术加速补助金和犹他大学眼科预防失明研究无限制补助金支持。作者感谢他的实验室过去和现在的成员在过去十年的杰出工作,Andrea Blitzer, M.D.在最初的文献综述和博士的帮助。Ralph Nelson (NIH)和Helga Kolb(犹他大学)提供有用的意见。
关于作者

David Krizaj在斯洛文尼亚卢布尔雅那大学获得学士学位,在纽约大学获得生理学和生物物理学博士学位,在加州大学旧金山分校完成视网膜神经生物学博士后研究。他在犹他大学医学院的莫兰眼科中心担任职务,在那里他拥有眼科的约翰弗雷德里克卡特捐赠教授职位。Krizaj博士的兴趣包括细胞内信号通路,专注于钙稳态、突触传递和机械转导,在过去的5年里,他的实验室一直积极从事机械敏感色氨酸TRP、链孔和压电通道在眼压调节中的作用的研究。另一个主要目标是确定小梁网细胞、视网膜神经元和胶质细胞的压力敏感性的分子机制,以及其调节失调如何导致糖尿病视网膜病变和青光眼。联系克里扎吉医生david.krizaj@hsc.utah.edu.
参考文献
- 弗拉克斯曼,s.r., R.R.伯恩,S.雷斯尼科夫,P.阿克兰,T.布雷斯韦特,M.V.西西内利,A.达斯,J. b .乔纳斯,J.基夫,J. h .肯彭,1990-2020年失明和远处视力障碍的全球原因:系统综述和荟萃分析《柳叶刀全球卫生》,2017;5 (12): e1221-e1234。[PubMed]
- Ko,研究。, D.-K。黄,W.-T。陈,c c。李和C.J.刘,社会经济地位对原发性开角型青光眼和闭角型青光眼诊断的影响:一项以台湾全国人口为基础的研究。PloS one。2016;11 (2): e0149698。[PubMed]
- 百老汇的察佐斯博士,青光眼史上的争议:这都是古老的希腊文吗?英国眼科杂志。2007;91(11): 1561 - 1562。[PubMed]
- 莱弗勒,c.t., S.G.施瓦茨,F.M.吉里伯蒂,M.T.杨,D.贝穆德斯,20世纪以前青光眼叫什么?眼科学和眼病。2015;7:牛津英语词典。S32004。[PubMed]
- 麦肯齐,W。a。休森和t。W。琼斯,眼科疾病实用专著.1855年:布兰查德和丽娅。[PubMed]
- 罩,提高我们对青光眼损伤的理解和检测:一种基于光学相干断层扫描(OCT)的方法。视网膜与眼睛研究进展。2017;57:46 - 75。[PubMed]
- 史密斯,C., J. Vianna和B. Chauhan,评估视网膜神经节细胞损伤。眼睛。2017;31(2): 209。[PubMed]
- 哈沃斯,r.s., M.克劳福德,L.J.弗里斯曼,S.维斯瓦纳坦,E.L.史密斯三世和L.卡特-道森,实验性青光眼的视野缺损和神经损失。视网膜与眼睛研究进展。2002;21(1): 91 - 125。[PubMed]
- 帕夫利迪斯,M., T.斯图普,R.纳斯卡尔,C.琴吉斯,灭霸,视网膜神经节细胞对晚期青光眼的抵抗:用碳菁染料DiI对人视网膜的尸检研究。调查眼科学和视觉科学。2003;44(12): 5196 - 5205。[PubMed]
- 尼克尔斯,r.w., G.R.豪厄尔,I.索托,还有S.W.约翰,压力下:青光眼期间的细胞和分子反应,青光眼是一种常见的神经退行性疾病和轴突病。神经科学年度回顾。2012;35:153 - 179。[PubMed]
- 利比,R.T, d·b·古尔德,m·g·安德森和s·w·约翰,青光眼易感性的复杂遗传。基因组学和人类遗传学年度综述。2005;6.[PubMed]
- 维格斯,J.L.和L.R.帕斯夸里,遗传学的青光眼。人类分子遗传学。2017;26日(R1): R21-R27。[PubMed]
- 乔纳斯,j。b。昂,r。r。伯恩,m布伦,R.里奇和S.熊猫-乔纳斯,青光眼。柳叶刀》。2017;390(10108): 2183 - 2193。[PubMed]
- 奎格利,H.A。,了解青光眼视神经病变:临床观察与调查的协同作用。视觉科学年度回顾。2016;2:235 - 254。[PubMed]
- 温雷布,R.N, T.昂和F.A.梅代罗斯,青光眼的病理生理和治疗进展。《美国医学协会杂志》上。2014;311(18): 1901 - 1911。[PubMed]
- 阿林厄姆,r.r., K.F.达吉,M.B.希尔兹,青光眼教材.2011年第6版,费城:Wolters Kluwer Health/Lippincott Williams & Wilkins。十四、610 p。
- 乔纳斯,j.b., P.韦伯,N.长冈,K.大野松井,高度近视和乳头旁三角洲区青光眼。《公共科学图书馆•综合》。2017;12 (4): e0175120。[PubMed]
- Tham,研究。,昂特,范琼,孙明明。Saw, R.G. Siantar, T.Y. Wong和c.y。程,眼压和近视对原发性开角型青光眼风险的联合作用:新加坡眼病流行病学研究。科学报告。2016;6:19320。[PubMed]
- arma, M。正常眼眼压的遗传决定。眼科档案。1967;78(2): 187 - 192。[PubMed]
- Khor、c.c.、T. Do、H. Jia、M. Nakano、R. George、K. Abu-Amero、R. Duvesh、l . j Chen、Z. Li和M. e . Nongpiur,全基因组关联研究发现原发性闭角型青光眼的5个新的易感位点。自然遗传学。2016;48(5): 556。[PubMed]
- 佐德、g.s.、K.E.布格、K.莫汉、S.D.格罗兹达尼奇、J.C.彼得斯、D.R.科恩、M.G.安德森、R.H.卡登、E.M.斯通和V.C.谢菲尔德,眼用4-苯基丁酸钠对原发性开角型青光眼心肌鼠模型的青光眼有拯救作用。调查眼科学和视觉科学。2012;53(3): 1557 - 1565。(PubMed)
- 峰岸,中山,户岛,川濑,岩田,视神经蛋白突变在青光眼和其他疾病中的意义。视网膜与眼睛研究进展。2016;55:149 - 181。(PubMed)
- 索雷夫松、G.B.沃尔特斯、A.W.翰威特、G.马森、A.赫尔加森、A.德万、A.西于尔兹松、A.约那斯多蒂尔、S.A.古德荣松和K.P.马格努松,CAV1和CAV2附近的常见变异与原发性开角型青光眼相关。自然遗传学。2010;42(10): 906。[PubMed]
- 麦格温、G.、S.麦克尼尔、C.奥斯利、C.吉金、D.爱泼斯坦和P.P.李,他汀类药物和其他降胆固醇药物与青光眼的存在。眼科的档案。2004;122(6): 822 - 826。[PubMed]
- 戴维斯、l.k.、K.J.迈耶、E.I.辛德勒、J.S.贝克、D.S.路德、A.J.格兰德斯塔德、T.E.谢茨、T.A.布劳恩、J.H.芬特和W.L.奥尔沃德,复制数变异与原发性开角型青光眼。调查眼科学和视觉科学。2011;52(10): 7122 - 7133。[PubMed]
- 布朗格,b.m., r。弗彻肖夫和e。r。塔姆,青光眼的房水流出通路:疾病机制和病因治疗的统一概念。欧洲制药与生物制药杂志,2015;95:173 - 181。[PubMed]
- 瑞斯坎普,地方检察官,助理检察官乔,上午弗莱,F.巴斯克斯-奇纳,N.麦考利,W.B.托雷森,D.克里扎伊,肿胀和类二十烷素代谢物在视网膜神经元和胶质细胞中差异地闸门TRPV4通道。神经科学杂志,2014;34(47): 15689 - 15700。[PubMed]
- 贝内特,t.m., D.S.麦凯,C.J.齐格弗里德,a。谢尔斯,三聚氰胺相关阳离子通道TRPM3的突变是遗传性白内障和青光眼的基础。PloS one。2014;9 (8): e104000。[PubMed]
- 利顿,p.b., p·冈萨雷斯和d·l·爱泼斯坦,蛋白水解细胞系统在小梁网稳态中的作用。实验研究。2009;88(4): 724 - 728。[PubMed]
- Rao, p.v., P.P. Pattabiraman和C. Kopczynski,Rho GTPase/Rho激酶信号通路在青光眼发病和治疗中的作用:从实验到床边的研究。实验研究。2017;158:23-32。[PubMed]
- Leske, m.c., A. Heijl, M. Hussein, B. Bengtsson, L. Hyman和E. Komaroff,青光眼进展的因素和治疗效果:早期显着青光眼试验。眼科的档案。2003;121(1): 48-56。[PubMed]
- 莫札法里耶,M.和J.弗拉默,对正常张力性青光眼发病机制及治疗的新认识。药理学最新观点。2013;13(1): 43-49。[PubMed]
- 芬格特、j.h.、K.米勒、A.赫德伯格-布恩茨、B.R.鲁斯、C.J.刘易斯、R.F.马林斯和M.G.安德森,转基因TBK1小鼠具有正常张力性青光眼的特征。人类分子遗传学。2016;26(1): 124 - 132。[PubMed]
- 卡玛尔,D.和R.希钦斯,正常紧张性青光眼——一种实用的方法。英国眼科学杂志。1998;82(7): 835 - 840。[PubMed]
- krizhaj, D., D.A. Ryskamp, N. Tian, G. Tezel, C.H. Mitchell, v.z Slepak和V.I. Shestopalov,从机械敏感性到炎症反应:青光眼病理的新参与者。当前研究。2014;39(2): 105 - 119。[PubMed]
- 克拉克,A.F.和R.J.沃丁格,类固醇在流出阻力中的作用。实验研究。2009;88(4): 752 - 759。[PubMed]
- 豪厄尔、g.r.、R.T.利比、T.C.雅各布斯、R.S.史密斯、F.C.法兰、J.W.巴特、J.M.巴贝、J.K.马尔尚、N.马赫什和V.波恰蒂,DBA/2J型青光眼早期视神经视网膜神经节细胞轴突受到损伤。细胞生物学杂志,2007;179(7): 1523 - 1537。[PubMed]
- 惠特莫,a.v., R.T.利比,还有S.W.约翰,青光眼:用新方式思考——自主轴突自我破坏和其他分隔过程的作用?视网膜与眼睛研究进展。2005;24(6): 639 - 662。[PubMed]
- 刘,B., S.麦克纳利,J.I.基尔帕特里克,S. p .贾维斯和C.J.奥布莱恩,青光眼的老化和眼组织僵硬。眼科学的调查。2018;63(1): 56 - 74。[PubMed]
- 伊佐蒂,M.朗戈巴迪,C.卡蒂利亚和S.C.萨卡,小梁网的线粒体损伤只发生在原发性开角型青光眼和假性剥脱型青光眼。《公共科学图书馆•综合》。2011;6 (1): e14567。[PubMed]
- 列夫科维奇-维宾,H. S.凡德,D.马卡洛夫斯基和F.拉文斯基,随着年龄的增长,视网膜神经节细胞对眼压升高的易感性增加,其内源性神经保护机制受损。分子的愿景。2013;19:2011。[PubMed]
- 阿科特、t.s.、M.J.凯利、K.E.凯勒、J.A.弗兰卡、D.W.阿布-哈桑、X.李、M.阿加和J.M.布拉德利,眼压内稳态:在高压环境中保持平衡。眼科药理学与治疗学杂志。2014;30(2 - 3): 94 - 101。[PubMed]
- 刘建华、张旭、D.F. Kripke和R.N. Weinreb,24小时眼压模式与早期青光眼改变相关。调查眼科学和视觉科学。2003;44(4): 1586 - 1590。[PubMed]
- Heijl, A., M.C. Leske, B. Bengtsson, L. Hyman, B. Bengtsson和M. Hussein,降低眼压和青光眼进展:来自早期明显青光眼试验的结果。眼科的档案。2002;120(10): 1268 - 1279。[PubMed]
- 克鲁克斯、K.R.、R.R.阿林厄姆、秦x、刘玉云、J.R.吉布森、C.圣地亚哥-图拉、K.R.拉罗克-艾布拉姆松、E.德尔波诺、P.查拉和L.W.赫恩登,原发性开角型青光眼的全基因组连锁扫描:祖先和诊断年龄的影响PloS one。2011;6 (7): e21967。[PubMed]
- 李鸿源波动,非人灵长类动物的眼压遥测。Exp Eye Res. 2015;141:91-8。[PubMed]
- 阿格尼菲利、L.、D.皮拉戈斯蒂诺、A.马斯特罗帕斯夸、V.法萨内拉、L.布雷西亚、G.M.托西、P.萨切塔和L.马斯特罗帕斯夸,原发性开角型青光眼的分子生物标志物:从无创到有创,卷221,在大脑研究的进展.2015年,爱思唯尔。p .学会年会。[PubMed]
- 凯恩斯,e。a。w。h。鲍德里奇,法医凯利,内源性大麻素系统作为青光眼的治疗靶点。神经可塑性。2016;2016.[PubMed]
- 斯塔默,W.D.和A.F.克拉克,小梁网状细胞的多个面。实验研究。2017;158:112 - 123。[PubMed]
- Ryskamp,地方检察官,点弗莱、T.T. Phuong、O. Yarishkin、A.O. Jo、Y. Xu、M. Lakk、A. Iuso、S.N. Redmon和B. Ambati,TRPV4调节哺乳动物眼内钙稳态、细胞骨骼重塑、常规流出和眼压。科学报告。2016;6:30583。[PubMed]
- 天鹤座,O.-J。, U. Grüsser-Cornehls, R. Kusel和A. Przybyszewski,视网膜神经节细胞对眼球变形的反应:“压力膦烯”的神经生理学基础。视觉研究。1989;29(2): 181 - 194。[PubMed]
- 孔,Y.X, J.G. Crowston, A.J. Vingrys, I.A. Trounce, B.V. Bui,小鼠急性眼压升高时及后视网膜的功能变化。调查眼科学和视觉科学。2009;50(12): 5732 - 5740。[PubMed]
- Ou, Y., R.E. Jo, E.M. Ullian, R.O. Wong, L. Della Santina,短暂性高眼压后特定视网膜神经节细胞类型和突触的选择性脆弱性。神经科学杂志,2016;36(35): 9240 - 9252。[PubMed]
- 德拉·桑蒂娜,L, D.M.英曼,C.B.卢皮恩,P.J.霍纳和R.O.黄,青光眼小鼠模型中不同类型视网膜神经节细胞结构和功能改变的差异进展。神经科学杂志,2013;33(44): 17444 - 17457。[PubMed]
- 孔、弗莱、萨马拉伊、吉尔伯特和斯坦库勒,全球儿童失明原因的进展和不断变化的流行病学的最新进展。美国儿童眼科和斜视协会杂志。2012;16(6): 501 - 507。[PubMed]
- 卡尔金斯,不论是青光眼神经退行性变进展的关键致病事件。视网膜与眼睛研究进展。2012;31日(6):702 - 719。[PubMed]
- El-Danaf, R.N.和A.D. Huberman,早期青光眼树突重塑的特征模式:来自基因鉴定的视网膜神经节细胞类型的证据。神经科学杂志,2015;35(6): 2329 - 2343。[PubMed]
- 冯亮、陈宏、易杰、J.B. Troy、张慧芳、刘欣,脑源性神经营养因子对高眼压小鼠视网膜神经节细胞和视觉功能的长期保护作用。调查眼科学和视觉科学。2016;57(8): 3793 - 3802。[PubMed]
- 威廉姆斯,p.a., J.R.特里布尔,K.W.佩珀,S.D.克罗斯,B.P.摩根,J.E.摩根,S.W.约翰,还有G.R.豪厄尔,抑制补体级联的经典通路可防止青光眼的早期树突和突触变性。分子神经退化。2016;11(1): 26。[PubMed]
- 卡尔金斯、d.j.、M.佩克尼、M.L.库珀、L.贝诺维茨、D.卡尔金斯、M.库珀、J.克劳斯顿、A.休伯曼、E.约翰逊和R.卢,青光眼视神经再生疗法的挑战。实验研究。2017;157:28-33。[PubMed]
- 贝洛斯基,B, e,巴贝托,m。p。科尔曼,k。r。马丁,WldS基因可以延缓大鼠青光眼模型的轴突变性,但不能延缓体细胞变性。欧洲神经科学杂志,2008;28(6): 1166 - 1179。[PubMed]
- 奎格利,h.a., R.M.桑切斯,G.R.邓克尔伯格,N.L. L 'Hernault, T.A. Baginski,慢性青光眼选择性损伤大视神经纤维。调查眼科学与视觉科学1987;28(6): 913 - 920。[PubMed]
- 摩根J.E。青光眼中的选择性细胞死亡:它真的发生了吗?英国眼科杂志。1994;78(11): 875。[PubMed]
- 雅各布斯,tc, r。t。利比,y。本,s。w。约翰,r。h。马斯兰德,DBA/2J小鼠视网膜神经节细胞变性是拓扑性的,而不是细胞类型特异性的。细胞生物学杂志,2005;171(2): 313 - 325。[PubMed]
- 冯良、赵颖、吉田明、陈浩、杨建峰、金t.s.、仓俊、J.B. Troy及刘欣,持续高眼压诱发小鼠视网膜神经节细胞的树突变性,这种树突变性取决于细胞的类型和位置。调查眼科学和视觉科学。2013;54(2): 1106 - 1117。[PubMed]
- 里斯纳,M.L., S.帕西尼,M.L.库珀,W.S.兰伯特,D.J.卡尔金斯,青光眼早期进展中视网膜神经节细胞兴奋性增强的轴生机制。美国国家科学院学报。2018;115 (10): E2393-E2402。[PubMed]
- Križaj, D。视网膜神经节细胞的多模态感觉整合,在视网膜退行性疾病.2016年,施普林格。p . 693 - 698。[PubMed]
- de Sevilla Müller, l.p., A. Sargoy, A.R. Rodriguez和N.C. Brecha,黑视素神经节细胞是大鼠视网膜对轴索损伤最有抵抗力的神经节细胞类型。《公共科学图书馆•综合》。2014;9 (3): e93274。[PubMed]
- Lakk, M., D. Young, J.M. Baumann, A.O. Jo, H. Hu, D. kriakia,多模态TRPV1和TRPV4传感器在小鼠视网膜神经节细胞亚群中共定位,但在功能上不相互作用。细胞神经科学前沿。2018;12.[PubMed]
- 黄w、J.B. Fileta、A. Dobberfuhl、T. Filippopolous、Guo y、G. Kwon及C.L. Grosskreutz,眼内压升高可引起钙调磷酸酶裂解,而钙调磷酸酶抑制可阻断实验性青光眼视网膜神经节细胞死亡。国家科学院学报,2005;102(34): 12242 - 12247。[PubMed]
- 黄玮、邢玮、d.r yskamp、C. Punzo和D. krizhaj,ryanodine钙释放通道在C57BL6和DBA/2J小鼠菌株中的定位和表型特异性表达。实验研究。2011;93(5): 700 - 709。[PubMed]
- 克罗格、蒋W.C.、费尔登、阮a、林J.H.,内质网应激和未折叠蛋白在眼健康和疾病中的反应。2月日报。2019;286(2): 399 - 412。[PubMed]
- 盖默,C·p·伍德,g·奇德洛和r·j·卡森,青光眼的神经保护:最新进展和临床翻译。临床与实验眼科。2019;47(1): 88 - 105。[PubMed]
- 黑尔,w.a., E. WoldeMussie, R.N. Weinreb, H. Ton, G. Ruiz, M. Wijono, B. Feldmann, L. Zangwill和L. Wheeler,美金刚胺治疗减少猴实验性青光眼相关变化的有效性和安全性,II:结构措施。调查眼科学和视觉科学。2004;45(8): 2640 - 2651。[PubMed]
- Rutar M., R. Natoli, R. Albarracin, K. Valter和J. Provis,670纳米光治疗减少视网膜变性后补体繁殖。J神经炎症。2012;9:257。[PubMed]
- 林、j.h.a、B.K. Stafford、阮P.L.、连B.V.、王志强、K. Zukor、Z. He、ad . Huberman、神经活动促进成人视网膜轴突的远距离、靶向性再生。自然神经科学。2016;19日(8):1073。[PubMed]
- 杨,H., J.雷诺,H.洛克伍德,G.威廉姆斯,C.哈丁,L.雷耶斯,C.斯托维尔,S.K.加德纳和C.F.伯格因,猴眼青光眼拔罐结缔组织表型的临床和研究意义。视网膜与眼睛研究进展。2017;59:1-52。[PubMed]
- 埃拉吉,j·g·弗拉纳根,c·A·西蒙斯和c·r·埃瑟,巩膜硬度特性对视神经头生物力学的影响。生物医学工程年鉴。2010;38(4): 1586 - 1592。[PubMed]
- 伯戈因,C.F.和J.C.唐斯,前提和预测:视神经头生物力学是老年视神经头易感性和临床行为的基础。青光眼杂志》上。2008;17(4): 318。[PubMed]
- 豪厄尔、g.r.、索托、朱欣、M.瑞安、D.G. Macalinao、G.L.索萨、L.B.卡德尔、K.H. MacNicoll、J.M. Barbay和V. Porciatti,在青光眼小鼠模型中,放射治疗抑制单核细胞进入视神经头并防止神经损伤。临床调查杂志。2012;122(4): 1246 - 1261。[PubMed]
- 奥斯本,N.N, C. Núñez-Álvarez, B. Joglar和S. del Olmo-Aguado,青光眼:关注线粒体与发病机制和神经保护的关系。欧洲药理学杂志。2016;787:127 - 133。[PubMed]
- 邓格勒-克里什,c.m., M.A.史密斯,D.M.英曼,G.N.威尔逊,J.W.杨,S.D.克里什,在DBA/2J小鼠青光眼模型的视觉投射中,顺行转运障碍先于逆行转运障碍。神经科学前沿。2014;8:290。[PubMed]
- Bosco, M.R. Steele和M.L. Vetter,慢性青光眼小鼠模型的早期小胶质细胞激活。比较神经病学杂志。2011;519(4): 599 - 620。[PubMed]
- 英曼,D.M.和P.J.霍纳,反应性非增生性胶质细胞增生在慢性青光眼小鼠模型中占主导地位。神经胶质。2007;55(9): 942 - 953。[PubMed]
- 孙,D., M. Lye‐Barthel, R.H. Masland, T.C. Jakobs,小鼠视神经头星形胶质细胞的形态和空间排列。比较神经病学杂志。2009;516(1): -。[PubMed]
- Ly, T., N. Gupta, R.N. Weinreb, P.L. Kaufman, Y.H. Yücel,灵长类青光眼中外侧膝状核的树突可塑性。视觉研究。2011;51(2): 243 - 250。[PubMed]
- 古普塔,G.格林伯格,L.N. De Tilly, B. Gray, M. polemiditis, Y.H. Yücel,磁共振成像检测青光眼中膝状外侧核萎缩。英国眼科杂志。2009;93(1): 56 -。[PubMed]
- 吉拉茨,E. M.克拉斯,E.德凯斯特,M.萨利纳斯-纳瓦罗,L. De Groef, C. Van den Haute, I. Scheyltjens, V. Baekelandt, L. Arckens和L. Moons,光遗传刺激上丘在小鼠青光眼模型中给予视网膜神经保护。神经科学杂志,2019;39(12): 2313 - 2325。[PubMed]
- Coulombre有好处,A.J,。发育中的眼睛的细胞学,卷11,在细胞学国际评论.1961年,爱思唯尔。p . 161 - 194。[PubMed]
- 谢尔顿,L., D. Troilo, M.R. Lerner, Y. Gusev, D.J.布兰克特,J.S. Rada,绒毛膜/RPE基因表达在绒猴眼生长和屈光变化中的微阵列分析。分子的愿景。2008;14:1465。[PubMed]
- 施耐德,M.和R. Fuchshofer,星形胶质细胞在青光眼视神经头纤维化中的作用。实验研究。2016;142:49-55。[PubMed]
- Tamm e和C. Ethier,星形胶质细胞和青光眼神经退行性变参与者的Lasker/IRRF倡议。青光眼轴突损伤的生物学方面:简要综述。Exp Eye Res. 2017;157:5-12。[PubMed]
- t.c.手跟前,青光眼的差异基因表达。冷泉港医学观点。2014;4 (7): a020636。[PubMed]
- 托瓦尔·维达尔斯,R.J.沃丁格,A.F.克拉克,人眼视神经头部筛板细胞的鉴别与定位。实验研究。2016;147:94 - 97。[PubMed]
- H.曼苏尔、J.R.麦考姆、L.科尔、M.韦布二世、A.科尔林比尼斯和T.赞灵,大鼠视网膜星形胶质细胞间隙连接斑块中连接蛋白30的表达和连接蛋白异质性频率随年龄增加而增加。PloS one。2013;8 (3): e57038。[PubMed]
- 纽曼,弹性轴,神经胶质细胞通过释放神经递质来调节视网膜中的神经元活动和血流。《英国皇家学会哲学学报B辑:生物科学》2015;370(1672): 20140195。[PubMed]
- 埃尔南德斯、M.R。青光眼视神经头:星形胶质细胞在组织重塑中的作用。视网膜与眼睛研究进展。2000;19(3): 297 - 321。[PubMed]
- leye - barthel M., D. Sun和T.C. Jakobs,青光眼视神经星形胶质细胞的形态。调查眼科学和视觉科学。2013;54(2): 909 - 917。[PubMed]
- Dvoriantchikova G.和D. Ivanov,肿瘤坏死因子‐alpha介导NF的激活‐κB和JNK信号在视网膜神经节细胞和星形胶质细胞中以相反的方式级联。欧洲神经科学杂志,2014;40(8): 3171 - 3178。[PubMed]
- Johnson, e.c., L. Jia, W.O. Cepurna, T.A. Doser, J.C. Morrison,大鼠青光眼模型眼压升高后视神经头基因表达的全局变化。调查眼科学和视觉科学。2007;48(7): 3161 - 3177。[PubMed]
- 泰泽尔,G, mr . Hernandez和M.B. Wax,反应性星形胶质细胞迁移的体外评价,这是青光眼视神经头组织重塑的组成部分。神经胶质。2001;34(3): 178 - 189。[PubMed]
- 雷蒙,S., O. Yarishkin, M. Lakk和D. Krizaj,视网膜胶质细胞对机械敏感通道TRPV4的激活有不同的反应。调查眼科学与视觉科学。2018;59(9): 3939 - 3939。
- 钟,R.S.和K.R.马丁,神经胶质细胞相互作用与青光眼。目前眼科的观点。2015;26(2): 73。[PubMed]
- 瓦克斯,M.B.和G.泰泽尔,青光眼视网膜神经节细胞命运的免疫调节。实验眼睛研究。2009;88(4): 825 - 830。[PubMed]
- Anderson, s.r., J. Zhang, M.R. Steele, C.O. Romero, A.G. Kautzman, D.P. Schafer, M.L. Vetter,补体靶向新生视网膜神经节细胞,通过小胶质细胞进行吞噬消除。神经科学杂志,2019;39(11): 2025 - 2040。[PubMed]
- 布林,k.t., S.R.安德森,M.R.斯蒂尔,D.J.卡尔金斯,a.j.博斯克,M.L.维特,在慢性青光眼模型中,fractalkine信号的丢失加剧轴突运输功能障碍。神经科学前沿。2016;10:526。[PubMed]
- 王明、王旭、赵亮、马文豪、I.R. Rodriguez、R.N. Fariss及王炜泰,大胶质细胞-小胶质细胞通过TSPO信号相互作用调节小鼠视网膜小胶质细胞激活。神经科学杂志,2014;34(10): 3793 - 3806。[PubMed]
- Bosco, C.O. Romero, K.T. Breen, A.A. Chagovetz, M.R. Steele, B.K. Ambati和M.L. Vetter,通过观察慢性青光眼小鼠模型体内早期小胶质细胞的改变,可以预测神经退行性变的严重程度。疾病模型和机制。2015;8(5): 443 - 455。[PubMed]
- 卡尔斯泰特,M., R.肖尔茨,M. Rutar, W.T. Wong, J.M. Provis和T. Langmann,视网膜小胶质细胞:只是旁观者还是治疗目标?视网膜与眼睛研究进展。2015;45:30-57。[PubMed]
- 松本,H., S. Sugio, F. Seghers, D. Krizaj, H.秋山,Y.石崎,P. gly,和K.柴崎,视网膜脱离诱导的Müller胶质细胞肿胀激活TRPV4离子通道并在体温下触发感光细胞死亡。神经科学杂志,2018;38(41): 8745 - 8758。[PubMed]
- 莱辛巴赫,a。a。布林曼,Müller健康和病变视网膜中的细胞.科学与商业媒体。2010,纽约:施普林格Verlag。[PubMed]
- Jo, a.o., d.r yskamp, T.T. Phuong, A.S. Verkman, O. Yarishkin, N. MacAulay, D. kriaj,TRPV4和AQP4通道协同调节视网膜Müller胶质细胞体积和钙稳态。神经科学杂志,2015;35(39): 13525 - 13537。[PubMed]
- naawashiro, H., A. Messing, N. Azzam和M. Brenner,缺乏GFAP的小鼠对外伤性脑脊髓损伤敏感。Neuroreport。1998;9(8): 1691 - 1696。[PubMed]
- 莫里森,j.c., w.o.c.y Guo和E.C. Johnson,人青光眼视神经损伤的病理生理学:来自青光眼啮齿动物模型的见解。实验研究。2011;93(2): 156 - 164。[PubMed]
- 巴尔加斯,j.l.c., I.K.奥斯瓦尔德,n·恩塞恩,M.R.奥卢梭,P.A.巴克,d·鲍伊,a·迪·波罗,可溶性肿瘤坏死因子α通过钙透性AMPA受体激活促进青光眼视网膜神经节细胞死亡。神经科学杂志,2015;35(35): 12088 - 12102。[PubMed]
- 贾西姆,A.H.和D.M.英曼,高眼压模型中低氧胶质细胞的证据。调查眼科学与视觉科学,2019;60(1): 1 - 15。[PubMed]
- Jo, a.o., M. Lakk, A.M.弗莱、T.T.芳、S.N.雷蒙、R.罗伯茨、B.A.伯科维茨、O.雅瑞什金和D.克里扎伊,两种纤体细胞类型的不同容量调节和钙信号通路由TRPV4通道辅助。美国国家科学院学报,2016;113(14): 3885 - 3890。[PubMed]
- Križaj, D。没有细胞是孤岛:小梁网离子通道作为周围环境的传感器,卷3,在青光眼研究与临床进展J.R. Samples和P.A. Knepper,编辑,2019年,Kugler出版社阿姆斯特丹。
- 林奎斯特,N., Q. Liu, J. Zajadacz, K. Franze和A. Reichenbach,视网膜胶质细胞(Müller):感知和响应组织拉伸。调查眼科学和视觉科学。2010;51(3): 1683 - 1690。[PubMed]
- Niittykoski, M, G. Kalesnykas, K.P. Larsson, K. kararniranta, K. e . Åkerman和H. Uusitalo,青光眼实验模型中钙信号的改变。调查眼科学和视觉科学。2010;51(12): 6387 - 6393。[PubMed]
- Ribas, v.t., J.C. Koch, U. Michel, M. Bähr, P. Lingor,钙通道抑制剂对轴突变性的抑制作用可改善视神经挤压后视网膜神经节细胞存活和再生。分子神经生物学。2017;54(1): 72 - 86。[PubMed]
- 因格贝尔,丧张拉整体:细胞机械转导的架构基础。生理学年度回顾。1997;59(1): 575 - 599。[PubMed]
- Ranade, s.s., R. Syeda和A. Patapoutian,机械激活离子通道。神经元。2015;87(6): 1162 - 1179。[PubMed]
- Jaalouk, D.E.和J. Lammerding转导调控机制。Nature评论分子细胞生物学。2009;10(1): 63。[PubMed]
- 考克斯,c.d., N.巴维,B.马蒂纳克,力的起源:应用于压电通道的脂质力原理Volume79,膜的最新话题.2017年,爱思唯尔。59 - 96页。[PubMed]
- Phuong, t.t., S.N. Redmon, O. Yarishkin, J.M. Winter, D.Y. Li和D. krijaj,钙通过TRPV4通道流入调节视网膜微血管内皮细胞之间的粘附接触。生理学杂志。2017;595(22): 6869 - 6885。[PubMed]
- 萨平顿,r.m., T. Sidorova, N.J. Ward, R. Chakravarthy, K.W. Ho和D.J. Calkins,瞬时受体电位香草素-1 (TRPV1)的激活影响视网膜神经节细胞神经元对压力相关应激的反应。渠道。2015;9(2): 102 - 113。[PubMed]
- 雅瑞什金,O, T.T. Phuong, C.A. Bretz, K.W. Olsen, J.M. Baumann, M. Lakk, A. Crandall, C. Heurteaux, M.E. Hartnett和D. krijaj,TREK-1通道调节小梁网细胞的压力敏感性和钙信号。普通生理学杂志。2018;150(12): 1660 - 1675。[PubMed]
- 雷蒙,s.n,柴崎和D.克里扎伊,瞬时受体电位离子通道亚族V成员4,在信号分子百科全书,第二版主编,2017,施普林格出版社。
- 怀特,j·p, m·奇贝利,l·厄本,b·尼利乌斯,j·g·麦乔翁,i·纳吉,TRPV4:多元乐团的分子指挥。生理上的评论。2016;96(3): 911 - 973。[PubMed]
- Lapajne, L., M. Lakk, L. Gubeljak, O. Yarishkin, M. Hawlina, D. krijaj,角膜机械转导驱动trpv4依赖的传递蛋白ATP的释放。调查眼科学和视觉科学。2019;60岁,5 . .
- 瑞斯坎普,P.威特科夫斯基,P.巴拉巴斯,黄伟,C.克勒,N.P.阿基莫夫,李舜臣,S.乔汉,邢炜华,R.C. Rentería,多模态离子通道瞬时受体电位香草素4可调节小鼠视网膜神经节细胞的钙通量、峰值率和凋亡。神经科学杂志,2011;31日(19):7089 - 7101。[PubMed]
- 阿雷东多,D.Z, R.I. Noguez, A.C. Bautista, O.R. Vázquez, M. Bernardini, A.P. Fiorio, D. Gkika, N. Prevarskaya, F. Lopez-Casillas和W. Liedtke,TRPV4拮抗剂在血管抑制素对血视网膜屏障通透性的调节作用中的双重贡献:糖尿病环境起作用。科学报告。2017;7(1): 13094 - 13094。[PubMed]
- Lakk, M., O. Yarishkin, J.M. Baumann, A. Iuso, D. krizhaaj,胆固醇调节Müller胶质细胞的多模态感觉转导。神经胶质。2017;65(12): 2038 - 2050。[PubMed]
- Alloui, A., K. Zimmermann, J. Mamet, F. Duprat, J. Noel, J. Chemin, N. Guy, N. Blondeau, N. Voilley, C. Rubat‐Coudert,长途跋涉‐1,一个参与多模态疼痛感知的K+通道。EMBO杂志。2006;25(11): 2368 - 2376。[PubMed]
- 哈林顿,d·m·德雷克,视野:临床视野文献和图谱。6日.1990年,处于:费城。52-60页。
- 博诺米,L., G. Marchini, M. Marraffa和R. Morbio确定人群中眼压与青光眼之间的关系。Ophthalmologica。2001;215(1):品种马非常。[PubMed]
- 维奇诺,E, F.D. Rodriguez, N. Ruzafa, X. Pereiro和S.C. Sharma,哺乳动物视网膜中的胶质细胞-神经元相互作用。视网膜与眼睛研究进展。2016;51:1-40。[PubMed]
- 塞缪尔斯,b.c., J.T.西格沃特,詹威,L.赫斯考克斯,M.奇曼托,R.惠特利,J.C.唐斯和C.A.吉金,一种新的树鼩青光眼模型。调查眼科学和视觉科学。2018;59(7): 3136 - 3143。[PubMed]
- 塔姆,e.r., C.R.埃瑟,J.E.道林,c.s.唐斯,M.H.埃利斯曼,s.s Fisher, B. Fortune, M. Fruttiger, T. Jakobs, G. Lewis,青光眼轴突损伤的生物学方面:简要综述。实验研究。2017;157:5-12。[PubMed]
- Lieff, J。寻找心灵.2013;可以从:http://jonlieffmd.com/blog/are-microglia-the-most-intelligent-brain-cells.(PubMed)
- 莫兰,m.m., M.A.麦卡亚历山大,T. Bíró,还有A.萨拉西,瞬时受体电位通道作为治疗靶点。《自然》回顾了药物发现。2011;10(8): 601。[PubMed]
- 帕特尔,A.J.和E. Honoré,哺乳动物2P结构域K+通道的性质及调控。神经科学的趋势。2001;24(6): 339 - 346。[PubMed]
- 雅瑞什金,O., T.T. Phuong, M. Lakk和D. krijaj,TRPV4不调节视网膜远端光反应,在视网膜退行性疾病.2018年,施普林格。p . 553 - 560。[PubMed]